Role of drug metabolic enzymes and transporters in drug-drug interactions between antiretroviral and antituberculosis drugs
Abstract
Drug-drug interactions is one of the major determinant in drug development and clinical applications. Individual differences in pharmacokinetics may cause extensive variability in drug efficacy, toxicity and adverse drug reactions, and represent a major concern in drug development. According to world health organization, an estimated 1.2 million patients were diagnosed with HIV/TB coinfection in 2015, and approximately 400,000 coinfected individuals were died in 2016. During treatment of tuberculosis and human immunodeficiency virus (HIV) coinfections, a combination of multiple therapeutic drugs is used, which is potential for interactions between coadministered drugs. Drug metabolizing enzymes and transporters plays vital role in the disposition, pharmacokinetics as well as efficacy and toxicity of many antituberculosis (anti-TB) and antiretroviral (ARVs) drugs. The current understanding of drug-drug interactions involving the first-line antituberculosis and antiretroviral (ARVs) drugs are reviewed and summarized in this article emphasizing the role of drug transporters and metabolic enzymes. It is found that several clinical drug-interactions are caused by the phase I and II metabolic enzymes (cytochrome-P-450s, CYPs, UDP-glucuronosyltransferases, UGTs) and uptake/ efflux transporters, particularly permeability glycoprotein, brest cancer resistance protein, multidrug resistance protein and solute carrier transporters). Overall, this study will be helpful to understand the drug-interactions between the ARV or anti-TB drug regimen or with concomitantly used drugs at a glance. By understanding the unique functions and characteristics of enzymes and transporters, it may use in the management of drug interactions in the treatment of HIV and TB.
Keywords
INTRODUCTION
To date, it is very common to alteration of drug’s clinical response in multiple drug regimen by another drug is defined as a drug-drug interactions (DDI). The alterations of response may result from the change in drug disposition which is believed to cause by induction or inhibition of transporters or metabolic enzymes and drug transporters play role in drug absorption, distribution, metabolism and excretion (ADME), those DDIs are known for pharmacokinetic interactions [1]. Drug transporters belonging to two major super-families of membrane-associated proteins, the solute carriers (SLCs), which primarily function as a cellular drug influx mechanism, and the ATP-binding cassette (ABC) transporters which in contrast, efflux drugs/ compounds from cellular targets, are known to play a vital role in overall drug disposition (i.e., drug distribution to target tissues/cells and drug clearance from the liver and kidney). These transporters are also thought to be contributor in pharmacokinetic drug-drug interactions, clinical response as well as toxicity [2][3]. On the other hand, drug metabolizing enzymes comprising of the Phase I oxidative enzymes (cytochrome-P-450s, CYPs) and the conjugation Phase II enzymes (sulfotransferases, glucuronyl transferases, N-acetyltransferases and glutathione-S-transferases), are key players in exogenous/ endogenous compound and drug metabolism, and are differentially expressed in various mammalian tissues. The pharmacokinetic DDIs are responsible for approximately 20-30% of the adverse drug reactions in general population, they also account for about 10% of the cases under emergency department and contribute 3-5% of the medication errors in-patients [4][5]. Since the drug pharmacokinetics can be significantly altered by both transporters and enzyme mediated DDIs, it can potentially affect to the therapeutic efficacy or toxicity of a drug [6]. Generally, DDIs based on metabolism are due to induction and/or inhibition of metabolic enzymes (CYP450s or UGTs), are considered as most prominent cause [7]. Based on the therapeutic and toxic effects of both the parent drug and its metabolites, the clinical consequences depends on the CYP450 enzyme induction or inhibition and this may be particularly significant to those drugs has narrow therapeutic window because DDIs based on metabolism may cause changes in the concentration of drug up to 10 fold whose biotransformation is induced or inhibited [8]. To date, drug-metabolizing enzymes mediated DDIs have been extensively studied in vitro as well as in vivo, emerging understanding has led to uptake and efflux transporters being recognized as a significant determinant of drug disposition that may cause significant DDIs. Passive diffusion through the membrane has been viewed as dominant in the disposition of most drugs, but it is now well recognized that carrier-mediated transport plays a vital role [9]
Acquired Immunodeficiency syndrome (AIDS) caused by Human Immunodeficiency Virus 1 (HIV-1) is considered as a major public health issue globally. In 2016, an estimated 1.2 million people died of AIDS-related illness out of 36.7 million people affected with this disease worldwide [10]. Although, the combination of antiretroviral therapy (cART) has effectively decreased HIV-1 related mortality and morbidity, some opportunistic infections are increasingly common and causing death of HIV patients. For example, tuberculosis (TB) is one of the leading opportunistic infections and cause of death in HIV patients. According to World Health Organization, an estimated 1.2 million patients were diagnosed with HIV/TB coinfection in 2015, and approximately 400,000 coinfected individuals were died in 2016 (World Health Organization, 2015, 2016b). In Europe, HIV/TB coinfection increased by 40% over the last 5 years [12]. Overall, the risk of developing TB is reported to be 26–31 times higher in HIV-infected compared to HIV non-infected persons [13]. Similarly, the most common adverse events such as psychiatric disorders (i.e., depression and anxiety) are found in HIV infected patients than non-infected. The prevalence of depression and anxiety were reported to be 22-50% and 2-40%, respectively in HIV infected individuals globally [14-15], whereas serious mental illness (SMI) in HIV infected individuals was estimated to be between 3% to 23%, a 10 fold higher occurrence than in the healthy population in United States (0.4%) [16-18].
The concurrent use of antiretroviral (ARV) and antituberculosis (anti-TB) drugs have significantly reduced the mortality rate of HIV/TB patients [19]. However, the use of multiple drug regimens in a combination are known to produce drug–drug interactions and overlapping toxicities which are the most challenging aspects of managing both diseases. According to recent guidelines, at least one ARV regimen from the integrase strand transfer inhibitors (INSTIs) class (Douletegravir/Elvitegravir/Raltegravir) and two from the nucleotide reverse transcriptase inhibitors (NRTIs) class (abacavir/lamivudine/tenofovir/emtricitabine) are recommended as a first line therapy for HIV infection (Table 1) [20]. In addition protease inhibitors (PIs) (Ritonavir/Atazanavir) are recommended as a boosting therapy depending on treatment outcomes [20]. Whereas, a combined regimen of rifampicin, isoniazid, ethambutol and pyrazinamide are recommended as a first line therapy for tuberculosis (TB) in the world [10]. HIV-infected individuals are often suffered with opportunistic infections, and are likely to be prescribed with multiple medications to manage complex medical conditions in addition to their ART regimens, making them vulnerable to the effects of potential and serious drug-drug interactions, caused by alteration in drug transport and metabolism affecting pharmacokinetic/pharmacodynamic (PK/PD) properties.
Table 1: Recommended antiretroviral regimen for initial therapy.
DISCUSSION
Drug Metabolizing Enzymes (DMEs)
Drug-metabolizing reactions are mainly classified into phase I and phase II metabolism. Briefly, phase I reactions involve oxidation, reduction or hydrolysis of the drug, and are primarily mediated by the cytochrome P450 (CYP450) family of enzymes. Phase II reactions involve covalently binding of an endogenous compound, most often glucuronide acid, glutathione or sulphate, to the phase I metabolite. For example, phase II enzymes are uridine diphosphoglucuronosyl transferases (UGTs) and glutathione-S-transferases (GSTs). The phase II conjugation produces a more polar metabolite and promotes excretion of the drug from the tissue, normally via efflux transporters. The CYP450 enzymes responsible for metabolism of xenobiotics are primarily expressed in the liver and intestines, and to less extend in the lung, kidney and central nervous system. CYP450 enzymes are generally located in the endoplasmic reticulum of cells where they carry out the nicotinamide adenine dinucleotide phosphate-oxidase (NADPH)-dependent oxidation of a diversity of substrates. The superfamily of CYP450 enzymes comprises 57 genes which have been organized into families (denoted by the first identification number, e.g. CYP3) and subfamilies (denoted by letters, e.g. CYP3A) [21]. The individual isoenzymes within each subfamily are further denoted by numbers, e.g. CYP3A4. CYP450 enzymes are in general promiscuous in their capacity to bind and metabolite substrates, and thus, there is significant overlapping substrate specificity among CYP enzymes. At present, among the human CYP450 isoenzymes, CYP1A1, CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4 and CYP3A5 are considered of particular importance in drug metabolism (Table 2) [22]. To date, it is hypothesized that the biotransformation of approximately 40-50% of all xenobiotic drugs on the market are caused by CYP3A-mediated oxidation [23]. For the majority of individuals, the human CYP3A isoform CYP3A4 are the most abundant CYP enzyme expressed in the liver and intestine, and considered as the major drug-metabolizing enzyme in human [24],[25].
Table 2. Major Drug Metabolizing Human CytochromeP450 Enzymes in Human.
Drug Transporters (DTs)
At present drug transporters are mainly classified into two major gene superfamily, the solute carrier (SLC) superfamily and the ATP-binding cassette (ABC) superfamily [9]. The members of the SLC superfamily are facilitated transporters or ion-coupled secondary transporters, frequently associated with uptake of compounds from the blood into tissues or organs such as the liver and kidney, or in the absorption from the gastrointestinal tract into the systemic circulation [26]. The SLC superfamily include 48 subfamilies of which the most investigated are the organic anion transporting polypeptides (OATPs, SLCO), the organic cation transporters (OCTs) and organic anion transporters (OATs), as well as the more recently identified multidrug and toxin extrusion (MATE, SLC47A) family which is involved in drug excretion from the kidney and liver (Table 3) [27].
Table 3. Major ADMET Drug Transporters in Human.
Interactions of ARVs and/ or Anti-TB Drugs with Membrane Transporters and Metabolic Enzymes
Several first line ARV therapy drugs, such as integrase strand transfer inhibitors (INSTIs) (raltegravir, dolutegravir and elvitegravir), nucleoside reverse transcriptase inhibitors (NRTIs) (lamivudine, abacavir, tenofovir, emtricitabine) and protease inhibitors (PIs) (atazanavir, ritonavir) are substrates of ABC efflux transporters such as P-glycoprotein (P-gp); multidrug resistance proteins (MRPs) and/or breast cancer resistance protein (BCRP), and thus could potentially interfere with the ABC transporter function of effluxing drugs from their cellular targets. In addition, metabolic enzymes such as members of the CYP450 family (e.g., CYP3A4) and UDP-Glucuronosyl transferase family (e.g., UGT1A1) can also contribute to an extensive intracellular degradation of several ARVs, particularly protease inhibitors and integrase strand transfer inhibitors [28]. Together, these effects can significantly contribute to poor tissue and intracellular ARVs concentrations [29]. Recently, this has been clearly demonstrated the atazanavir disposition is significantly involved with transporters in a P-gp/BCRP knockout mouse [30]. In particular, significantly higher atazanavir brain and testicular tissue concentrations was found among the P-gp knockout compared to the wild type mice suggesting that the efflux transporters localized at the blood-brain barrier and blood-testicular barrier are important determinants of this HIV protease inhibitor tissue distribution [30]. In addition, several anti-TB drugs are known to be substrates and potent modulators of drug transporters (influx and efflux) and metabolic enzymes (both phase I and II enzymes), and are implicated in many clinical drug- interactions [31-33]. For example, among the first line anti-TB drug regimen, rifampicin is known as a substrate, inducer and inhibitor of liver enzymes (CYP450s & UGTs), and P-gp, OATPs (OATP1B1, -1B3) and implicated in the reduction of many concomitant drugs (statins) [31],[34],[35],[36]. Similarly, ethambutol is another first line anti-TB drug undergoes non-cyp450 mediated metabolism and substrate/inhibitor of multiple OCT transporters [37],[38],[39].
Complexities of Drug-Drug Interactions Involving Anti-HIV and Anti-TB/Psychotic Drugs
The combination of HIV and TB often is referred to as a syndemic, where two or more diseases interact in a synergistic fashion, and leads to an overall increased burden of both diseases [40]. One associated factor for such occurrence could the use of multiple drug regimens for the treatment of HIV co-infection with TB, and the overlapping drug-drug interactions (DDI) involving drug transport and metabolism, which may result in toxic and/or sub-therapeutic drug levels leading to therapy change or treatment failure. Several DDIs involving anti-HIV and anti-TB drugs have been reported previously. For example, in healthy volunteers raltegravir AUC was increased by 77% when coadministered with rifapentine and this DDIs was poorly understood [41]. On the other hand, rifampin (rifampicin analogue) decreased raltegravir concentration by up to 60% among healthy subjects [42]. It is known that, raltegravir primary metabolize by UGT1A1 and actively transported by P-gp and BCRP could play role in this DDIs. Another integrase inhibitor, Dolutegravir is primarily metabolized by UGT1A1 and CYP3A4 (lesser extent) [43] and also a potent inhibitor of OCT2 as well as substrate of P-gp and BCRP [44]. In a phase-1 clinical study, rifampin was reported to decrease dolutegravir AUC approximately 54% whereas rifabutin showed no significant effect on dolutegravir pharmacokinetics [45]. However, dolutegravir is a potent inhibitor of organic cation transporter 2 (OCT2) and multidrug and toxic compound extrusion (MATE) transporters, and metformin is transported by OCTs transporter for disposition. As a result, metformin higher plasma concentration was found when coadministered with dolutegravir among healthy subjects. Regarding the first line therapy ARV drugs, particularly NRTI that has been studied with raltegravir is the tenofovir. In a study among healthy subjects, the plasma concentrations of raltegravir was increased by 49% due to a yet unexplained mechanism which needs further investigation to understand the exact mechanism of this clinical interactions [46]. In another study, raltegravir concentrations was reported to increase by 72% when combinedly administered with atazanavir in healthy subjects [47]. Similarly, dolutegravir AUC was increased approximately 91% and 62% (when boosted with ritonavir) by atazanavir in healthy subjects [43]. Whereas, atazanavir is known to inhibit UGT1A1 enzyme, P-gp and BCRP efflux transporters [48] responsible for metabolism and transport of raltegravir and dolutegravir [49][44]. It was proposed that, inhibition of enzymes and transporters by atazanavir could play role in the substantial increase of raltegravir/dolutegravir plasma concentration. In addition, atazanavir is known to increase tenofovir AUC by 21% in healthy subjects [50]. The MRP2, a renal efflux pump could play role in this drug interactions which potentially could lead to renal toxicity [51]. This potential MRP2 mediated drug interactions remains unclear and further investigations would be helpful to understand. These complexity in drug interactions can be overcome by extensive mechanistic data for metabolizing enzymes and transporters.
Overlapping Toxicities with ARVs and Anti-TB Drug Regimen
Overlapping toxicities and adverse reactions are common when multiple ARVs and anti-TB drugs are co-administered in a given time [52] (Table 4) (Figure 1). For example, Isoniazid, stauvudine and didanosine are well-known for drug induced neuropathy [53]. As a result, increased hepatoxicity when nonnucleoside reverse transcriptase inhibitors, (NNRTIs, particularly nevirapine) were prescribed with anti-TB regimens was remains as a concerns for adverse event [54]. In another study, found an increased hepatotoxicity in efavirenz [54],[55] and nevirapine [54] co-administration with anti-TB regimens in 30% HIV/TB patients [56]. Again, in case of lopinavir [57], atazanavir [58] and saquinavir [59] boosting with ritonavir in combination with rifampin caused severe hepatotoxicity in healthy subjects. It seemed, most common hepatotoxicity was reported among the subjects when protease inhibitors (PI) boosted with rifampin. In regard to sequence of drug therapy may play a role and hepatoxic metabolite of PIs comes from the rifampin induced CYP450s or ritonavir may cause reduction in metabolic clearance was hypothesized[58].
Table 4. Overlapping ping side effect profiles of first-line antituberculosis drugs and antiretroviral drugs.
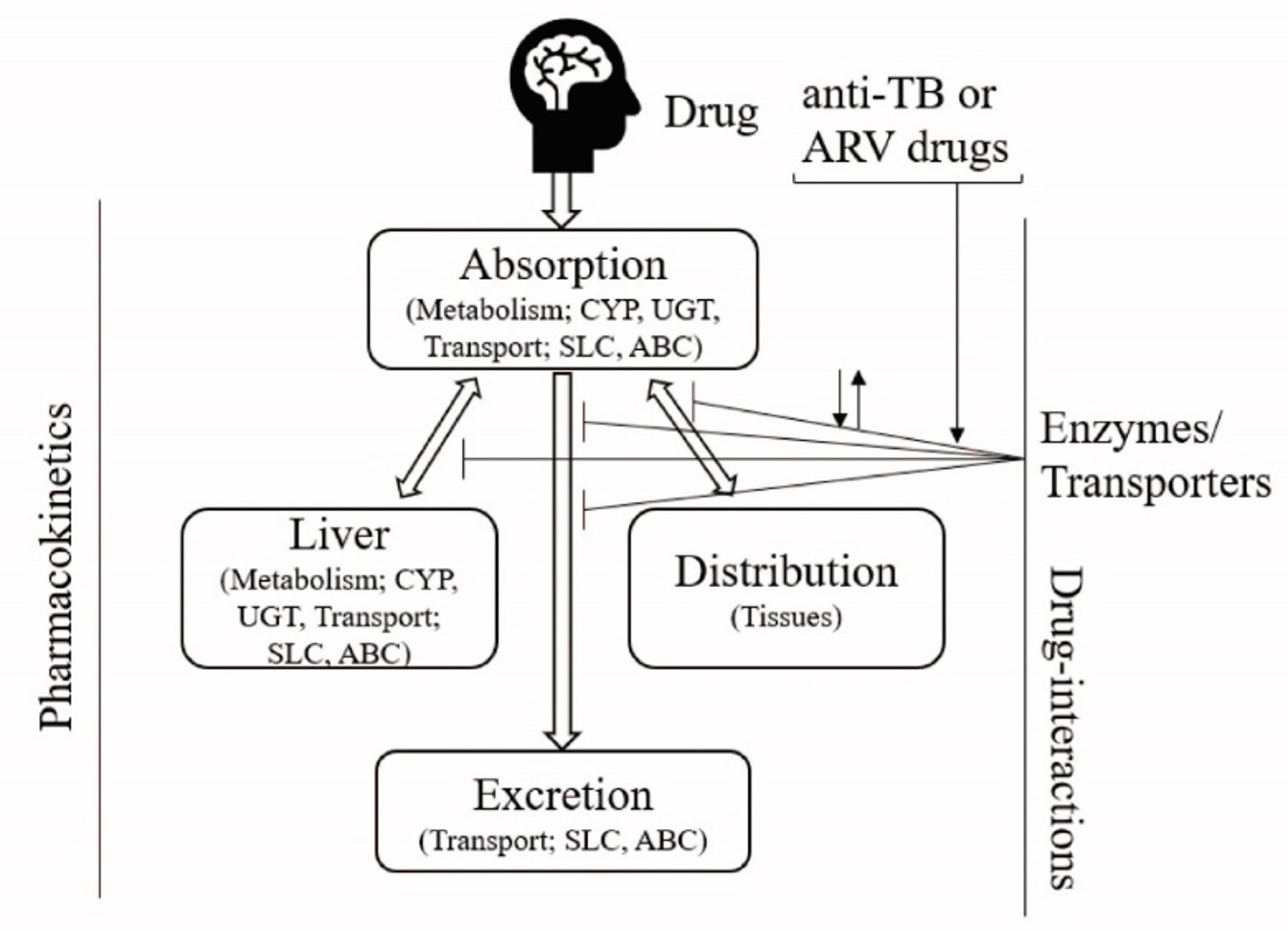
Transporter-Enzyme Interplay in Drug Disposition
At this advance science of age, a considerable overlap in substrate specificity and tissue distribution among membrane transporters and metabolizing enzymes were found throughout the body [60]. Recently, a significant substrate overlap between hepatic uptake transporters and enzymes has been recognized, [60] e.g. between CYP3A4 and OATPs [61]. However, the interactive nature of CYP3A and multidrug resistance protein 1 (MDR1) is the most extensively studied interplay between enzymes and transporters [60],[62],[63]. CYP3A and MDR1 act as a coordinated barrier for xenobiotics. [64] However, several clinical studies explained that the role of intestinal MDR1 greatly played role by limiting absorption of the drug [62],[63],[64]. In the intestine, where the drug enters the enterocytes from the luminal side, MDR1 majorly play role in the process of the drug reaching to the enzyme through repeated cycles of absorption and efflux, giving CYP3A mediated metabolism prior to enter into systemic circulation. This phenomenon may explained as the drug from the enterocytes return to the lumen without metabolism by P-gp mediated efflux or other apical efflux transporters thereby allowing re-entry into enterocytes and increased chance of metabolic conversion by CYP3A [65]. Again, in the liver, the drug enters into the hepatocytes from the basolateral side and encounters CYP3A prior to MDR1-mediated efflux into the bile canaliculi, drugs will not re-enter the cells because of concentration gradient, as result limited metabolism occurs and more parent drug cross the membrane.
CONCLUSIONS
Drug-drug interactions is common phenomena in multidrug regimen, especially in TB/ HIV treatment. The metabolic enzymes and transport proteins are differentially expressed in several tissues and play a crucial role in the drugs disposition and clinical response. The emphasis on metabolic enzymes and drugs transporters are well given since a couple of years and has been attracted by many to investigate on them for their clinical drug-interactions. After having several clinical mechanistic studies, it is believed that, several drugs are influenced by in the interplay between metabolic enzymes and drug transporters. Also, still limited data are available to conclude or characterize precisely the DDIs, there is a requirement of more mechanistic understanding of several complex drug-interactions between antituberculosis and antiretroviral drugs. This indicates the requirement of more mechanistic studies to understand clinical pharmacokinetic/ pharmacodynamic DDIs. Furthermore, base-poke studies on drug metabolic enzymes and transporters role in the disposition of the drug/s will be helpful to optimize drug-dose regimen in pharmacotherapy to HIV-TB.
ACKNOWLEDGEMENT
We are thankful to Md. Tozammel Hoque for excellent opinion on scientific aspects of this article. We declare no conflicts of interest or other relevant affiliations, financial involvement, or agreement/interest with any organization or governing body in this study. Additionally, there was no technical assistance in preparation of this manuscript.
AUTHOR CONTRIBUTIONS
SS performed the experiment; MNI and MEH conceived the study; SS and MNI analyzed the data; SS and MNI. wrote the paper.
CONFLICT OF INTERESTS
The authors declare no conflict of interest.
References
- [1]Köhler GI, Bode-Böger SM, Busse R, Hoopmann M, Welte T, Böger RH. Drug-drug interactions in medical patients: effects of in-hospital treatment and relation to multiple drug use. Int J Clin Pharmacol Ther. 2000; 38: 504–13.
- [2]König J, Müller F, Fromm MF. Transporters and Drug-Drug Interactions: Important Determinants of Drug Disposition and Effects. Pharmacol Rev. 2013; 65: 944-66.
- [3]Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013; 138: 103–41.
- [4]Hohl CM, Dankoff J, Colacone A, Afilalo M. Polypharmacy, adverse drug-related events, and potential adverse drug interactions in elderly patients presenting to an emergency department. Ann Emerg Med. 2001; 38: 666–71.
- [5]Leape LL, Bates DW, Cullen DJ, Cooper J, Demonaco HJ, Gallivan T et al. Systems analysis of adverse drug events. ADE Prevention Study Group. Jama. 1995; 274: 35–43.
- [6]Leape LL, Bates DW, Cullen DJ, Cooper J, Demonaco HJ, Gallivan T et al. Systems analysis of adverse drug events. ADE Prevention Study Group. Jama. 1995; 274: 35–43.
- [7]Pirmohamed M. Cytochrome P450 enzyme polymorphisms and adverse drug reactions. Toxicology. 2003; 192: 23–32.
- [8]Teo YL, Ho HK, Chan A. Metabolism-related pharmacokinetic drug-drug interactions with tyrosine kinase inhibitors: Current understanding, challenges and recommendations. Br J Clin Pharmacol. 2015; 79: 241–53.
- [9]Consortium IT, Giacomini K, Huang S-M, Tweedie D, Benet L, Brouwer K et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010; 9: 215–36.
- [10]World Health Organization. Global Health Observatory (GHO) data: Global situation and trends of HIV. 2016; http://www.who.int/gho/hiv/en/.
- [11]World Health Organization. Tuberculosis and HIV. 2016; http://www.who.int/hiv/topics/tb/about_tb/en/.
- [12]Gao J, Zheng P, Fu H. Prevalence of TB/HIV Co-Infection in Countries Except China: A Systematic Review and Meta-Analysis. PLoS One. 2013; 8.
- [13]World Health Organization (WHO). Tuberculosis and HIV. http://www.who.int/hiv/topics/tb/about_tb/en/.
- [14]C O-LBSLSEA. HIV infection and psychiatric illness. African J Psychiatry (South Africa). 2009; 12: 115–28.
- [15]Del Guerra FB, Fonseca JLI, Figueiredo VM, Ziff EB, Konkiewitz EC. Human immunodeficiency virus-associated depression: Contributions of immuno-inflammatory, monoaminergic, neurodegenerative, and neurotrophic pathways. J. Neurovirol. 2013; 19: 314–27.
- [16]Lee S, Rothbard AB, Noll E, Blank MB. Use of HIV and psychotropic medications among persons with serious mental illness and HIV/AIDS. Adm Policy Ment Health. 2011; 38: 335–44.
- [17]Cournos F, McKinnon K. HIV seroprevalence among people with severe mental illness in the United States: A critical review. Clin. Psychol. Rev. 1997; 17: 259–69.
- [18]Rabkin JG. HIV and depression: 2008 Review and update. Curr. HIV/AIDS Rep. 2008; 5: 163–71.
- [19]Abdool Karim SS, Naidoo K, Grobler A, Padayatchi N, Baxter C, Gray AL et al. Integration of antiretroviral therapy with tuberculosis treatment. N Engl J Med. 2011; 365: 1492–501.
- [20]AIDS info. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents Living with HIV. 2017.
- [21]Nelson DR, Zeldin DC, Hoffman SMG, Maltais LJ, Wain HM, Nebert DW. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics. 2004; 14: 1–18.
- [22]Guengerich FP, Martin M V., Beaune PH, Kremers P, Wolff T, Waxman DJ. Characterization of rat and human liver microsomal cytochrome P-450 forms involved in nifedipine oxidation, a prototype for genetic polymorphism in oxidative drug metabolism. J Biol Chem. 1986; 261: 5051–60.
- [23]Thummel KE, Wilkinson GR. In vitro and in vivo drug interactions involving human CYP3A. Annu Rev Pharmacol Toxicol. 1998; 38: 389–30.
- [24]Guengerich FP. Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu Rev Pharmacol Toxicol. 1999; 39: 1–17.
- [25]Paine MF, Hart HL, Ludington SS, Haining RL, Rettie AE, Zeldin DC. The human intestinal cytochrome P450 ‘pie’. Drug Metab Dispos. 2006; 34: 880–86.
- [26]Lin L, Yee SW, Kim RB, Giacomini KM. SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug Discov. 2015; 14: 543–60.
- [27]Roth M, Obaidat A, Hagenbuch B. OATPs, OATs and OCTs: The organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br. J. Pharmacol. 2012; 165: 1260–87.
- [28]Kis O, Sankaran-Walters S, Hoque MT, Walmsley SL, Dandekar S, Bendayan R. HIV-1 Alters Intestinal Expression of Drug Transporters and Metabolic Enzymes: Implications for Antiretroviral Drug Disposition. Antimicrob Agents Chemother. 2016; 60: 2771–81.
- [29]Alam C, Whyte-Allman SK, Omeragic A, Bendayan R. Role and modulation of drug transporters in HIV-1 therapy. Adv Drug Deliv Rev 2016; 103: 121–43.
- [30]Robillard KR, Chan GNY, Zhang G, la Porte C, Cameron W, Bendayan R. Role of P-Glycoprotein in the Distribution of the HIV Protease Inhibitor Atazanavir in the Brain and Male Genital Tract. Antimicrob Agents Chemother. 2014; 58: 1713–22.
- [31]Shimokawa Y, Yoda N, Kondo S, Yamamura Y, Takiguchi Y, Umehara K. Inhibitory Potential of Twenty Five Anti-tuberculosis Drugs on CYP Activities in Human Liver Microsomes. Biol Pharm Bull. 2015; 38: 1425–29.
- [32]Cao L, Greenblatt DJ, Kwara A. Inhibitory Effects of Selected Antituberculosis Drugs on Common Human Hepatic Cytochrome P450 and UDP-glucuronosyltransferase Enzymes. Drug Metab Dispos. 2017; 45: 1035–43.
- [33]Te Brake LHM, Russel FGM, Van Den Heuvel JJMW, De Knegt GJ, De Steenwinkel JE, Burger DM et al. Inhibitory potential of tuberculosis drugs on ATP-binding cassette drug transporters. Tuberculosis. 2016; 96: 150–57.
- [34]Bolt HM. Rifampicin, A Keystone Inducer of Drug Metabolism: From Herbert Remmer’s Pioneering Ideas to Modern Concepts. Drug Metab Rev. 2004; 36: 497–09.
- [35]Glaeser H, Drescher S, Eichelbaum M, Fromm MF. Influence of rifampicin on the expression and function of human intestinal cytochrome P450 enzymes. Br J Clin Pharmacol. 2005; 59.
- [36]Ma X, Cheung C, Krausz KW, Shah YM, Wang T, Idle JR et al. A double transgenic mouse model expressing human pregnane X receptor and cytochrome P450 3A4. Drug Metab Dispos. 2008; 36: 2506–12.
- [37]Te Brake LHM, Van Den Heuvel JJMW, Buaben AO, Van Crevel R, Bilos A, Russel FG et al. Moxifloxacin is a potent in vitro inhibitor of OCT- and MATE-mediated transport of metformin and ethambutol. Antimicrob Agents Chemother. 2016; 60: 7105–114.
- [38]Pan X, Wang L, Gründemann D, Sweet DH. Interaction of ethambutol with human organic cation transporters of the SLC22 family indicates potential for drug-drug interactions during antituberculosis therapy. Antimicrob Agents Chemother. 2013; 57: 5053–59.
- [39]Parvez MM, Kaisar N, Shin HJ, Jung JA, Shin JG. Inhibitory Interaction Potential of 22 Antituberculosis Drugs on Organic Anion and Cation Transporter of SLC22A family. Antimicrob Agents Chemother. 2016; 60: 6558–67.
- [40]Kwan C, Ernst JD. HIV and tuberculosis: A deadly human syndemic. Clin. Microbiol. Rev. 2011; 24: 351–76.
- [41]Weiner M, Egelund EF, Engle M, Kiser M, Prihoda TJ, Gelfond JAL et al. Pharmacokinetic interaction of rifapentine and raltegravir in healthy volunteers. J Antimicrob Chemother. 2014; 69: 1079–85.
- [42]Wenning LA, Hanley WD, Brainard DM, Petry AS, Ghosh K, Jin B et al. Effect of rifampin, a potent inducer of drug-metabolizing enzymes, on the pharmacokinetics of raltegravir. Antimicrob Agents Chemother. 2009; 53: 2852–56.
- [43]Song I, Borland J, Chen S, Lou Y, Peppercorn A, Wajima T et al. Effect of atazanavir and atazanavir/ritonavir on the pharmacokinetics of the next-generation HIV integrase inhibitor, S/GSK1349572. Br J Clin Pharmacol. 2011; 72: 103–08.
- [44]Reese MJ, Savina PM, Generaux GT, Tracey H, Humphreys JE, Kanaoka E et al. In vitro investigations into the roles of drug transporters and metabolizing enzymes in the disposition and drug interactions of dolutegravir, a hiv integrase inhibitor. Drug Metab Dispos. 2013; 41: 353–61.
- [45]Dooley KE, Sayre P, Borland J, Purdy E, Chen S, Song I et al. Safety, tolerability, and pharmacokinetics of the HIV integrase inhibitor dolutegravir given twice daily with rifampin or once daily with rifabutin: results of a phase 1 study among healthy subjects. J Acquir Immune Defic Syndr. 2013; 62: 21–27.
- [46]Wenning LA, Friedman EJ, Kost JT, Breidinger SA, Stek JE, Lasseter KC et al. Lack of a significant drug interaction between raltegravir and tenofovir. Antimicrob Agents Chemother. 2008; 52: 3253–58.
- [47]Iwamoto M, Wenning LA, Mistry GC, Petry AS, Liou SY, Ghosh K et al. Atazanavir modestly increases plasma levels of raltegravir in healthy subjects. Clin Infect Dis 2008; 47: 137–40.
- [48]Weiss J, Rose J, Storch CH, Ketabi-Kiyanvash N, Sauer A, Haefeli WE et al. Modulation of human BCRP (ABCG2) activity by anti-HIV drugs. J Antimicrob Chemother. 2007; 59: 238–45.
- [49]Hoque MT, Kis O, De Rosa MF, Bendayan R. Raltegravir permeability across blood-tissue barriers and the potential role of drug efflux transporters. Antimicrob Agents Chemother. 2015; 59: 2572–82.
- [50]Hoetelmans RMW, Mariën K, De Pauw M, Hill A, Peeters M, Sekar V et al. Pharmacokinetic interaction between TMC114/ritonavir and tenofovir disoproxil fumarate in healthy volunteers. Br J Clin Pharmacol. 2007; 64: 655–61.
- [51]Tourret J, Deray G, Isnard-Bagnis C. Tenofovir Effect on the Kidneys of HIV-Infected Patients: A Double-Edged Sword? J Am Soc Nephrol. 2013; 24: 1519–27.
- [52]Perriëns JH, St Louis ME, Mukadi YB, Brown C, Prignot J, Pouthier F et al. Pulmonary tuberculosis in HIV-infected patients in Zaire. A controlled trial of treatment for either 6 or 12 months. N Engl J Med. 1995; 332: 779–84.
- [53]McIlleron H, Meintjes G, Burman WJ, Maartens G. Complications of antiretroviral therapy in patients with tuberculosis: drug interactions, toxicity, and immune reconstitution inflammatory syndrome. J Infect Dis 2007; 196 Suppl: S63-75.
- [54]Shipton LK, Wester CW, Stock S, Ndwapi N, Gaolathe T, Thior I et al. Safety and efficacy of nevirapine- and efavirenz-based antiretroviral treatment in adults treated for TB-HIV co-infection in Botswana. Int J Tuberc Lung Dis. 2009; 13: 360–66.
- [55]La Porte CJL, Colbers EPH, Bertz R, Voncken DS, Wikstrom K, Boeree MJ et al. Pharmacokinetics of Adjusted-Dose Lopinavir-Ritonavir Combined with Rifampin in Healthy Volunteers. Antimicrob Agents Chemother. 2004; 48: 1553–60.
- [56]Yimer G, Ueda N, Habtewold A, Amogne W, Suda A, Riedel KD et al. Pharmacogenetic & pharmacokinetic biomarker for efavirenz based ARV and rifampicin based anti-TB drug induced liver injury in TB-HIV infected patients. PLoS One 2011; 6.
- [57]Nijland HMJ, L’homme RFA, Rongen GA, van Uden P, van Crevel R, Boeree MJ et al. High incidence of adverse events in healthy volunteers receiving rifampicin and adjusted doses of lopinavir/ritonavir tablets. AIDS. 2008; 22: 931–35.
- [58]W HD, L KS, Laura L, A KM, Carol S, G GJ et al. Hepatotoxicity and Gastrointestinal Intolerance When Healthy Volunteers Taking Rifampin Add Twice-Daily Atazanavir and Ritonavir. JAIDS-JOURNAL Acquir IMMUNE Defic Syndr 2009; 50: 290–93.
- [59]Schmitt C, Riek M, Winters K, Schutz M, Grange S. Unexpected hepatotoxicity of rifampin and saquinavir/ritonavir in healthy male volunteers. Arch Drug Inf. 2009; 2: 8–16.
- [60]Benet LZ. The drug transporter-metabolism alliance: Uncovering and defining the interplay. Mol. Pharm. 2009; 6: 1631–43.
- [61]Karlgren M, Ahlin G, Bergström CAS, Svensson R, Palm J, Artursson P. In Vitro and in silico strategies to identify OATP1B1 Inhibitors and Predict Clinical Drug-Drug Interactions. Pharm Res. 2012; 29: 411–26.
- [62]Benet LZ, Cummins CL. The drug efflux-metabolism alliance: Biochemical aspects. In: Advanced Drug Delivery Reviews. 2001 doi:10.1016/S0169-409X(01)00178-8.
- [63]Wacher VJ, Wu CY, Benet LZ. Overlapping substrate specificities and tissue distribution of cytochrome p450 3A and P-glycoprotein: Implications for drug delivery and activity in cancer chemotherapy. Mol. Carcinog. 1995; 13: 129–34.
- [64]Zhang Y, Benet LZ. The gut as a barrier to drug absorption: combined role of cytochrome P450 3A and P-glycoprotein. Clin Pharmacokinet. 2001; 40: 159–68.
- [65]Lown KS, Mayo RR, Leichtman AB, Hsiao HL, Turgeon DK, Schmiedlin-Ren P et al. Role of intestinal P-glycoprotein (mdr1) in interpatient variation in the oral bioavailability of cyclosporine. Clin Pharmacol Ther. 1997; 62: 248–60.