A reliable and easy method to isolate a pure population of bovine granulosa cells from slaughterhouse ovaries for in vitro studies
Abstract
In a growing follicle, granulosa cells (GCs) are the major cellular component and perform several important functions. GCs are an excellent choice for in vitro experimentation to understand their functions in vivo. However, the isolation of a pure fraction of GCs from slaughterhouse ovaries remains a challenge. The current study describes a simpler yet reliable method for isolating GCs from slaughterhouse ovaries of domestic animals. For this, bovine ovaries were collected from a nearby slaughterhouse and GCs were collected by aspiration method, one fraction of fresh GCs was snap frozen and stored at -80°C for further analysis and another fraction was used in culture purpose. The purity of GCs was determined by quantifying follicle stimulating hormone receptor (FSHR) and CYP17A1 transcripts and analyzing the cultured cells under a microscope. The viability of GCs ranged from 66 – 35 % depending on the time and media used in transportation-processing. The results showed that FSHR gene was highly expressed as evidenced by the presence of a strong band while CYP17A1 is completely absent in the GCs isolated using the current method. In addition, the characteristics of the cultured cells further confirm the purity of isolated GCs. The current method isolates a pure population of GCs from slaughterhouse ovary without any contamination of thecal cells.
INTRODUCTION
Follicular development, also known as folliculogenesis, is a dynamic process which involved different morphological changes of follicular cells. The whole process is orchestrated by different molecules in a tightly organized manner [1,2]. A developmentally competent oocyte is released for fertilization at the end of follicular development. Among the cellular constituents, granulosa cells (GCs) are the major cell type in a follicular microenvironment. GCs are the somatic cells closely associated with the developing female gamete or oocyte. At the beginning of follicular development, GCs undergo a rapid proliferation and apoptosis in order to create a fluid-filled cavity called follicular antrum [3]. It also performs several other important functions including converting androgen to estradiol, the supply of nutrients to oocyte during growth, support and protects oocyte during ovulation. In addition, GCs contribute significantly contribute to the extracellular vesicles content of the follicular fluid, however, the function of these vesicles in follicular microenvironment has yet to be confirmed [4]. At the end of follicular development, after ovulation, GCs are transformed into granulosa-lutein cells by a preovulatory pituitary luteinizing hormone (LH) surge [5]. The transformation of GCs to granulosa-lutein cells is particularly very important for a successful ovulation and subsequent formation of corpus luteum to maintain the pregnancy. Therefore, it appears that the function and integrity of GCs are crucial for mammalian female fertility [6].
To understand and unmasking the functions of GCs, in vitro cell culture studies played a significant role. More than half a century ago GCs were cultured and proliferation was reported in both human and other experimental animals [7]. Since then many studies have conducted by isolating GCs from growing follicles to understand various aspects of GC function. We have been working on slaughterhouse originated bovine GCs for a long time and published several articles regarding the function and molecular aspects of GCs [4,5,8]. Researchers have tried different methods to collect GCs in their study. However, isolating a pure population of GCs from slaughterhouse ovaries using a simpler method remains a significant challenge. In this study, based on my previous experiences of isolation of GCs [4,5,8,9], I reported a comparatively easy and reliable method for GCs isolation from slaughterhouse originated bovine ovaries.
MATERIALS AND METHODS
Chemicals and reagents
Chemicals and reagents were purchased from Sigma-Aldrich unless otherwise stated.
Workflow
For a quick understanding of the isolation procedure of GCs from slaughterhouse ovaries, the whole workflow is presented in Fig. 1.
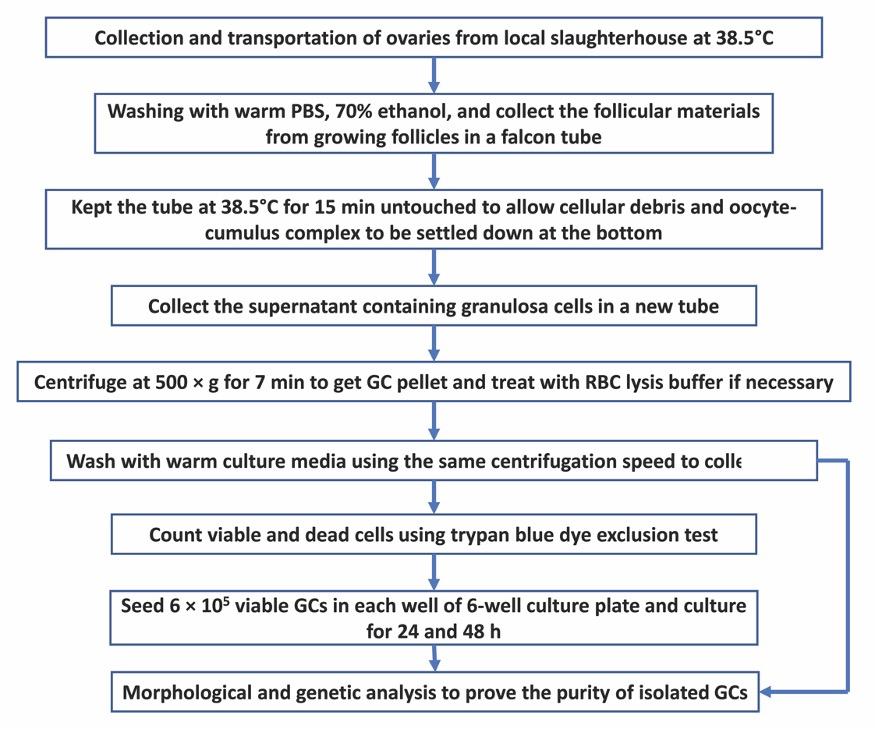
Collection of ovaries
A total of 30 ovaries were collected from 15 cattle (Bos Taurus) at random stages of the estrus cycle from a local slaughterhouse on a single visit. Collected ovaries were kept immediately in a thermos-flask containing collection medium (Tissue Culture Medium 199 + 100 μg/mL streptomycin and 100 U/mL penicillin) at 38.5°C and brought to the laboratory for processing. Ovaries were divided into three groups and each group consists of 10 ovaries. Ovaries were trimmed and washed twice with warm PBS to remove dirt. After that, ovaries were dipped in 70% ethanol for 30 seconds. Ovaries were washed twice with warm PBS and kept in collection medium at 38.5°C during processing.
Isolation of GCs
Follicular materials including GCs, oocyte-cumulus complex, and follicular fluid were collected from medium-sized follicles (4-8 mm diameter) using a 5 mL syringe with an 18 G needle attached to it. Follicular materials from different follicles were put in a 50 mL falcon tube containing 10 mL serum-free DMEM/F-12 with 1.25 mg/mL collagenase and 0.5 mg/mL DNase as previously described [10]. Collagenase and DNase were used to prevent cell clumping. In the case of collagenase and DNase unavailability, serum-free DPBS can also be used, however, it may result in lower cell viability. After collecting follicular materials from all ovaries, the falcon tubes containing follicular materials were thoroughly shaken and kept at 38.5°C for 15 min to allow cellular debris and cumulus-oocyte complex to be settled down at the bottom of the tube. The upper liquid fraction containing GCs were then carefully collected in a new 50 mL falcon tube and centrifuged at 500 × g for 7 min to obtain GCs pellet at the bottom of the falcon tube. To remove the red blood cells, GCs were resuspended 1-mL of red blood cell lysis buffer (8.26 g/L NH4Cl in 10 mM Tris-HCl) for 30 sec. Immediately 10 mL of DMEM/F-12 media containing 10% FBS (Pan-Biotech GmbH, Aidenbach, Germany), 100 μg/mL streptomycin and100 U/mL penicillin was added to restore the isotonicity. Following another wash with culture media, one fraction of cells was resuspended with 2 mL of culture media and kept at 38.5°C to use in cell culture and another fraction was kept at -80°C for further genetic analysis.
Cell counting and viability
Before seeding the cells, the viability of cells was determined using the trypan blue dye exclusion test as described previously [11] with few modifications. Briefly, 300 µl of DPBS, 100 µl trypan blue (0.4%) dye, and 100 µl of cell suspension were added to a microcentrifuge tube and mixed gently (dilution factor is 5) and incubated for 1-2 min. On the top of the counting chamber of a hemocytometer, a cover slide was placed and typically, 10-20 µl of trypan blue cell mix was placed each side of the hemocytometer. Under the microscope, live cells were appeared as clear (unstained) while dead cells appeared as blue (stained). Upon counting the cells in large chambers in four corners, the percentage of the viable cell was calculated as {%viable cells = (average number of viable cells/numbers of total cells) × 100}. Total number of viable cells in 1 mL was counted as follows- (Total number of viable cells/mL = average number of viable cells × dilution factor × 104).
Cell culture
Approximately 6 × 105 viable cells were seeded in each well of 6-well culture plate (Corning Incorporated, Kennebunk, ME, USA) and cultured in the culture media. Cells were cultured in a humidified environment of 5% CO2 at 38.5°C for different time-points. Culture media was changed at every 24 h time-point. Cells were routinely checked for their morphology and growth throughout the culture period using a phase-contrast light microscope (Nikon Eclipse TS100, Tokyo, Japan). Following the culture period, cells were washed twice with warm calcium-magnesium free PBS and detached by adding 700 µL 0.25% trypsin EDTA (Biological Industries, Kibbutz Beit Haemek, Israel) followed by an incubation at 38.5°C for 5-7 min. When all cells are detached from the floor, 3 mL of culture media was added to each well to stop the activity of trypsin EDTA. Cells were collected by centrifuging at 500 × g for 7 min and subjected to RNA isolation.
RNA isolation and cDNA synthesis
miRNeasy mini kit (Qiagen, Hilden, Germany) was used to isolate total RNA from GCs following the manufacturer’s protocol. Briefly, 700 µl of Qiazol lysis buffer was added and vortexed vigorously until all the cell clumps were dissolved and incubated for 6 min on the benchtop at room temperature. Following addition of 140 µl of chloroform, tubes were vigorously shaken for 15 s and centrifuged at 4°C for 15 min at 12,000 × g. The clear aqueous phase at the top was collected and transferred to a new 2-mL microcentrifuge tube and 1.5 volume of absolute ethanol (Merck KGaA, Darmstadt, Germany) was added and mixed by pipetting. RNA was captured in mini spin column’s membrane (provided by the company) and washed with washing buffer. In between the washing steps, residual DNA was removed by an on-column DNase digestion step using RNase-Free DNase set (Qiagen, Hilden, Germany). The concentration and integrity were determined using BioSpec-nano spectrophotometer (Shimadzu Biotech, Japan).
Maxima H Minus First Strand cDNA Synthesis Kit (Thermo Scientific, Massachusetts, USA) was used to synthesized complementary DNA from 1 µg of total RNA according to the previously described protocol [5]. Briefly, 1 μL 10 mM dNTP mix, 4 μL of 5× RT Buffer, 1 μL Maxima H Minus Enzyme mix, and 0.25μL oligo dT and random hexamer primers (each) was used in the cDNA synthesis reaction. First, in 1 µg of total RNA, dNTP mix, Random primer, and oligo dT was added and by adding nuclease-free water the reaction volume was adjusted to 15 μL and incubated at 65 °C for 5min. Following the incubation remaining components were added to the reaction mixture and incubated at room temperature (⁓ 25°C) for 10 min followed by 15 min at 50 °C. Finally, the reaction was terminated by heating at 85 °C for 5 min.
Polymerase chain reaction and gel documentation
PCR was performed using sequence-specific primers of three genes namely GAPDH (internal control), Cytochrome P450 17A1 (CYP17A1– theca specific), and Follicle Stimulating Hormone Receptor (FSHR– GC specific) were investigated for their expressions in GCs and theca cells. The sequence-specific primers were obtained from primer3web version 4.0.4 (http://bioinfo.ut.ee/primer3/) and the sequences are as follows- GAPDH- F 5´ CCAGGGCTGCTTTTAATTCT, R 5´- ATGGCCTTTCCATTGATGAC; FSH- F 5´- GCAGTCGAACTGAGGTTTGT, R 5´- AGATATCGGAGGTTGGGAAG; CYP17A1- F 5´- CTACTTGCCCTTTGGAGCAG, R 5´- GGAGTCATGAGGTGCTACCC. The PCR reaction was set up by adding 10 μl 2× Easy Taq PCR (Civic Bioscience, QC, Canada) master mix, 0.3 μM of each reverse and forward primers, 7.4 μL deionized water and 2 μL cDNA template. The thermal cycling conditions were 5 min at 95 °C followed by 35 cycles of denaturation at 95°C for 30 s, annealing at 56°C for GAPDH, 57°C for CYP17A1 55°C, and FSHR for 30 s, 72°C for 1 min for extension and at 72°C for 10 min for a final extension. The resulted PCR products were mixed with 6X loading dye (Thermo Scientific, Vilnius, Lithuania) and loaded into 2% agarose gel (Bio-Roc, Mainz, Germany) containing Ethidium bromide (EtBr) and visualized using Chemidoc XRS instrument (Bio-Rad, München, Germany).
RESULTS AND DISCUSSION
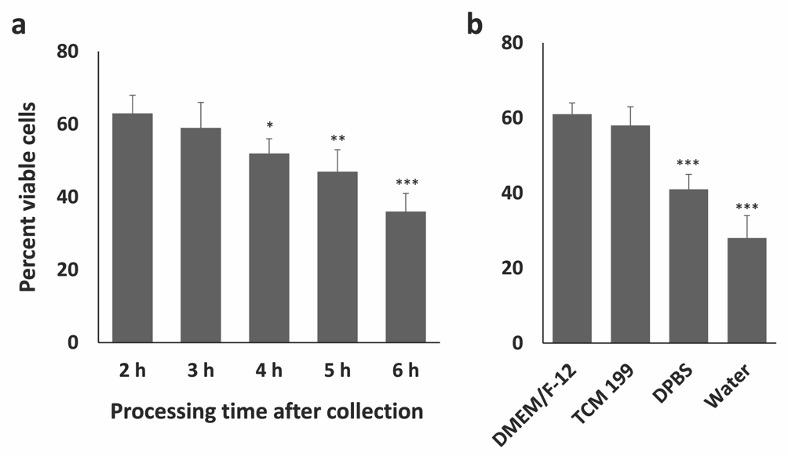
Collection of bovine ovaries from local slaughterhouse sometimes may involve long transportation time to the laboratory which may result in possible postmortem changes in the ovarian tissues and subsequently in the GCs. The duration of holding time, the temperature, and the media where the ovaries are held may potentially affect the outcome of an experiment. To understand these effects on bovine GCs, firstly, the impact of collection-transportation-processing time on the viability of GCs was measured. After the collection of cells, the viability of GCs was assessed using trypan blue exclusion test and the results showed that the viability ranges from 55-66% (Fig. 2) when the transportation-processing time is 2 h. A similar percentage of viable cells was previously obtained by [12] using a similar method of GC isolation. The viability of GCs crucially depends on the time spent in collection-transportation-isolation. In the current experiment, the GC viability was highest at 2 h time point (collection-transportation-processing) and the viability decreases as the time processing time increases. Significantly lower viability was obtained when the collection and transportation time is typically more than 4 h (Fig. 2b) and the lowest viability was observed in 6 h time point. Secondly, the effects of different types of collection media on the viability of GCs was assessed. It is important to note that the use of DPBS or water over collection media could also result in lower GC viability (35%) which has been shown in Fig. 2b, although the collection-transportation-processing time is less than 2 h. Therefore, the collection-processing time and collection media are crucial for obtaining a higher percentage of viable GCs. Apoptosis is a process of programmed cell death which may be responsible for the lower cell viability if the ovaries are maintained under suboptimal conditions for a long time. Indeed, the regulatory genes of apoptosis may be activated in the nucleus by the suboptimal environmental conditions including temperature, culture medium, and prolonged holding time could initiate the apoptosis process form the nucleus [13]. Apoptosis in GCs of feline ovaries significantly increased after 8h when stored in 4°C [14], while holding equine ovaries for 2-4 h after slaughter did not change the apoptosis level in GCs [15]. Interestingly, lower holding temperature may delay the apoptosis in granulosa cells as the enzymes are efficient at body temperature or higher [16]. However, the present study did not investigate the effect of holding temperature on the viability of GCs.
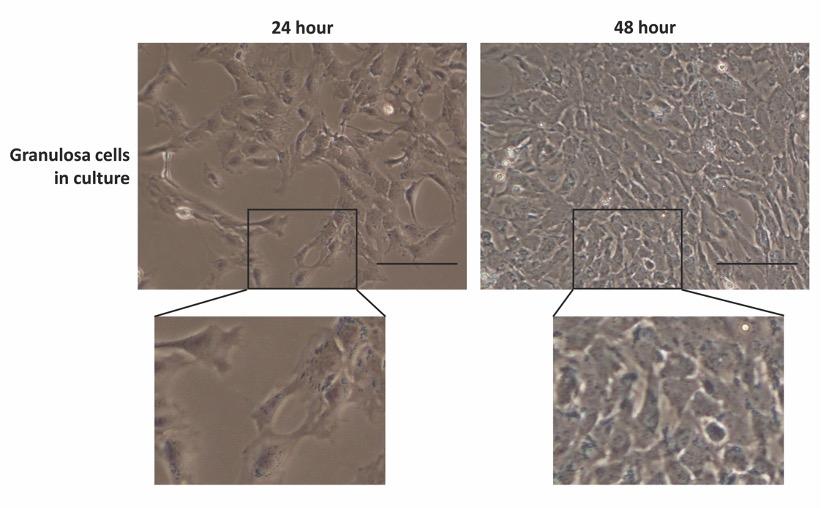
The shape and morphology of GCs in culture were evaluated using a phase-contrast microscopy and presented in Fig. 3. A total of 15 observations from each well of the 6-well plates were taken randomly and analyzed for possible theca cell contamination in the culture. The images revealed that GCs were apparently healthy and formed clusters of the flattened monolayer with typical epithelioid morphology. Similar morphology was previously described for healthy GCs in culture [17,18]. However, no theca cell (strip-like elongated cells) morphologies were observed in the cultured cells. The current GC isolation method efficiently recovered healthy GCs and avoid theca cell contamination during GC collection.
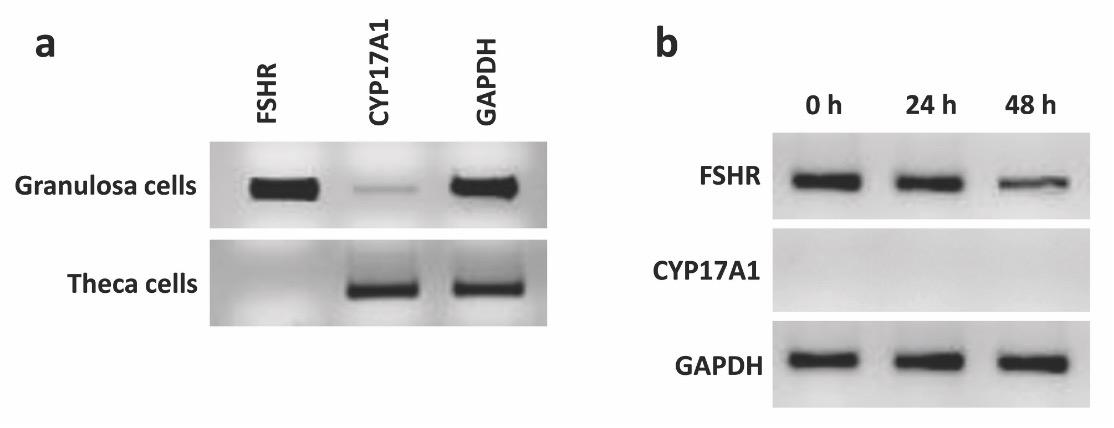
The expression of FSHR and CYP17A1 genes were checked for the presence and absence, respectively, in both freshly isolated and cultured GCs. FSHR gene is expressed highly in freshly isolated GCs while CYP17A1 predominantly expressed in theca cells and doesn’t express in GCs [19]. Therefore, the expression of these two genes was checked in GCs isolated using the current method. The results showed that FSHR was highly expressed in freshly isolated GCs and the expression is reduced in a time-dependent manner and the lowest expression was observed in 48 h cultured GCs (Fig. 4). FSHR mRNA is expressed in GCs obtained from all follicles. However, the expression is highest in the GCs of growing follicles, typically 6-8 mm in diameter, while the expression is lowest in small follicles and preovulatory follicles [20]. The GCs of the current experiment were collected from follicles ranged 4-8 mm in diameter, therefore, a strong expression of FSHR was seen in freshly isolated granulosa cells. The expression of FSHR mRNA in cultured hen GCs significantly decreased after 20 h [20] which is strongly support the findings of the current experiment as a time-dependent decrease was also seen in the cultured bovine GCs. Furthermore, no expression was observed for CYP17A1 in neither freshly isolated or cultured GCs at different time-points (Fig. 4).
CONCLUSION
The results of the current article confirm that the viability of granulosa cells crucially depends on both the transportation-processing time and the media used during transportation-isolation. Finally, considering all the factors, this article reported an easy and reliable method of isolating a pure population of GCs without theca cell contamination from slaughterhouse ovary that can be used further in vitro studies to understand GC biology.
CONFLICT OF INTEREST
The author declares that no conflict of interest exists.
References
- [1]Sohel MMH, Cinar MU. Advancement in Molecular Genetics to understand the molecular reproduction of Livestock – follicular development, Res. Agric. Livest. Fish. 2015; 1: 47–60. doi:10.3329/ralf.v1i1.22355.
- [2]Yada H, K. Hosokawa, Tajima K, Hasegawa Y, Kotsuji F. Role of ovarian theca and granulosa cell interaction in hormone productionand cell growth during the bovine follicular maturation process., Biol. Reprod. 1999; 61: 1480–6. http://www.ncbi.nlm.nih.gov/pubmed/10569992.
- [3]Matsuda F, Inoue N, Manabe N, Ohkura S. Follicular Growth and Atresia in Mammalian Ovaries: Regulation by Survival and Death of Granulosa Cells, J. Reprod. Dev. 2012; 58: 44–50. doi:10.1262/jrd.2011-012.
- [4]Sohel MMH, Hoelker M, Noferesti SS, Salilew-Wondim D, Tholen E, Looft C, et al., Exosomal and non-exosomal transport of extra-cellular microRNAs in follicular fluid: Implications for bovine oocyte developmental competence, PLoS One. 2013; 8. doi:10.1371/journal.pone.0078505.
- [5]Sohel MMH, Konca Y, Akyuz B, Arslan K, Sariozkan S, Cinar MU. Concentration dependent antioxidative and apoptotic effects of sulforaphane on bovine granulosa cells in vitro, Theriogenology. 2017; 97: 17–26. doi:10.1016/j.theriogenology.2017.04.015.
- [6]Tesfaye D, Salilew-Wondim D, Gebremedhn S, Sohel MMH, Pandey HO, Hoelker M, et al., Potential role of microRNAs in mammalian female fertility, Reprod. Fertil. Dev. 2017; 29. doi:10.1071/RD16266.
- [7]Mansani FE, Observations on the histogenesis of human granulosa cells cultured and proliferated in vitro., Quad. Clin. Ostet. Ginecol. 1959; 14: 630–47. http://www.ncbi.nlm.nih.gov/pubmed/14420735.
- [8]Sohel MMH, Cinar MU, Kalibar M, Arslan K, Sariozkan S, Akyuz B, et al., Appropriate concentration of Hydrogen Peroxide and Sulforaphane for granulosa cells to study oxidative stress in vitro, J. Biotechnol. 2016; 231: S24. doi:10.1016/j.jbiotec.2016.05.104.
- [9]Sohel MMH, Amin A, Prastowo S, Linares-Otoya L, Hoelker M, Schellander K, et al., Sulforaphane protects granulosa cells against oxidative stress via activation of NRF2-ARE pathway, Cell Tissue Res. 2018; 1–13. doi:10.1007/s00441-018-2877-z.
- [10]Langhout DJ, Spicer LJ, Geisert RD. Development of a culture system for bovine granulosa cells: effects of growth hormone, estradiol, and gonadotropins on cell proliferation, steroidogenesis, and protein synthesis., J. Anim. Sci. 1991; 69: 3321–34. http://www.ncbi.nlm.nih.gov/pubmed/1894569.
- [11]Strober W. Trypan blue exclusion test of cell viability., Curr. Protoc. Immunol. 2001; Appendix 3, Appendix 3B. doi:10.1002/0471142735.ima03bs21.
- [12]Feng T, Schütz LF, Morrell BC, Perego MC, Spicer LJ. Effects of N-carbamylglutamate and L-arginine on steroidogenesis and gene expression in bovine granulosa cells, Anim. Reprod. Sci. 2018; 188: 85–92. doi:10.1016/J.ANIREPROSCI.2017.11.012.
- [13]Tilly JL. Apoptosis and ovarian function. Rev. Reprod. 1996; 1: 162–72. http://www.ncbi.nlm.nih.gov/pubmed/9414454.
- [14]Jewgenow K, Wood TC, Wildt DE. DNA degradation in mural granulosa cells of non- and slightly atretic follicles of fresh and cold-stored domestic cat ovaries, Mol. Reprod. Dev. 1997; 48: 350–355. doi:10.1002/(SICI)1098-2795(199711)48:3<350::AID-MRD8>3.0.CO;2-Q.
- [15]Dell’Aquila ME, Albrizio M, Maritato F, Minoia P, Hinrichs K. Meiotic Competence of Equine Oocytes and Pronucleus Formation after Intracytoplasmic Sperm Injection (ICSI) as Related to Granulosa Cell Apoptosis1, Biol. Reprod. 2003; 68: 2065–2072. doi:10.1095/biolreprod.102.009852.
- [16]Pedersen HG, Watson ED, Telfer EE. Effect of ovary holding temperature and time on equine granulosa cell apoptosis, oocyte chromatin configuration and cumulus morphology, Theriogenology. 2004; 62: 468–480. doi:10.1016/J.THERIOGENOLOGY.2003.10.006.
- [17]Lawrence TS, Ginzberg RD, Gilula NB, Beers WH. Hormonally induced cell shape changes in cultured rat ovarian granulosa cells., J. Cell Biol. 1979. 80: 21–36. doi:10.1083/JCB.80.1.21.
- [18]Sangha GK, Kaur H, Khera KS. Morphological changes in cultured granulosa cells from different sized follicles of goat ovary, Indian J. Anim. Reprod. 2012;33. http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.879.6101&rep=rep1&type=pdf.
- [19]Nielsen ME, Rasmussen IA, Kristensen SG, Christensen ST, Mollgard K, Wreford Andersen E, et al., In human granulosa cells from small antral follicles, androgen receptor mRNA and androgen levels in follicular fluid correlate with FSH receptor mRNA, Mol. Hum. Reprod. 2011; 17: 63–70. doi:10.1093/molehr/gaq073.
- [20]Woods DC, Johnson AL. Regulation of Follicle-Stimulating Hormone-Receptor Messenger RNA in Hen Granulosa Cells Relative to Follicle Selection1, Biol. Reprod. 2005; 72: 643–650. doi:10.1095/biolreprod.104.033902.