Pathophysiological mechanisms of disease severity in COVID-19: An update
Abstract
The pandemic of COVID-19 has created a confounding global health crisis with more than 25 million cases and 870,000 deaths worldwide. The novel coronavirus, SARS-CoV-2, exploits the human ACE2 receptor and potential CD147 to invade a plethora of organ systems including cardiovascular, renal, endocrine, nervous, and gastrointestinal. The pathophysiological mechanisms of COVID-19 infection were found to be associated with the direct viral invasion, dysregulated renin-angiotensin-aldosterone system (RAAS), hypoxia, hyperinflammation, cytokine storm, endotheliopathy, and thrombosis. Emerging evidence suggests that the kinin-kallikrein system, iron dysregulation, and complement component C5a anaphylatoxin have roles in disease severity. In critical patients, the effects manifest as acute respiratory distress syndrome (ARDS), lymphopenia, acute kidney injury (AKI), disseminated intravascular coagulation (DIC), hypovolemic shock leading to multiorgan dysfunction syndrome (MODS). In this review, we provided an update on the pathophysiology of COVID-19 with an emphasis on the clinical outcomes in severe patients that will help facilitate a deeper understanding of the disease.
INTRODUCTION
In the matter of a decade, another human coronavirus of zoonotic origin emerged and put the world in an unprecedented crisis. Experiences from SARS (severe acute respiratory syndrome) in 2002, and MERS (Middle East respiratory syndrome) in 2012, led to the prediction of the emergence of a new respiratory disease, and COVID-19 (coronavirus disease-2019) prevailed as a pandemic. The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent for COVID-19, and it is the seventh coronavirus to infect humans [1]. The SARS-CoV-2 is a relatively large enveloped virus with an RNA genome size of about 30kb. Similar to SARS-CoV, the SARS-CoV-2 belongs to the lineage B of betacoronavirus and shares a 79% of genetic sequence similarity with SARS-CoV, and about 50% with MERS-CoV [2].
In most cases, COVID-19 causes asymptomatic to mild and moderate illness with fever, cough, dyspnea, and loss of smell although the disease can be life-threatening for critical patients [3]. A recent study has identified six categories of COVID-19 patients based on the clinical manifestations, of which three types of “severity” groups were observed [4]. The severe patients appeared to be elderly, overweight, and had pre-existing comorbidities like asthma or lung disease [4]. Similar to SARS and MERS, the COVID-19 causes viral pneumonia and acute respiratory distress syndrome (ARDS). The hyper inflammation and cytokine storm were previously reported in SARS, and similar immune responses were also observed in COVID-19 [5]. The novel coronavirus has claimed over 870,000 lives globally with an estimated infection fatality rate of 0.65% [6]. In contrast, the case fatality due to SARS was roughly 10%, whereas, for MERS, it was as high as 35% [7]. Studies show that SARS-CoV-2 can cause a plethora of clinical symptoms affecting systems such as cardiovascular, renal, endocrine, nervous, and gastrointestinal [8].
The current understanding of the various clinical outcomes in COVID-19 is yet in its early stage. However, since SARS-CoV-2 utilizes the same human ACE2 receptor as SARS-CoV and a high genetic similarity is present between two genomes, an analogous pattern of immune reaction is expected [9]. Exploring the pathogenesis and the associated pathophysiology of COVID-19 is fundamental as it bears relevance to both in the bench-side research and the bedside clinical applications. In this narrative review, we aimed to summarize the updated understanding and the advances in the molecular pathogenesis and the pathophysiology of COVID-19 with a focus on the clinical outcomes in severe patients, and discussed the course of disease progression in light of the existing literature.
ENTRY, ATTACHMENT, AND REPLICATION
SARS-CoV-2 primarily invades the lung cells and causes respiratory illness. It follows a typical lifecycle as of other human coronaviruses. Once inside the body, the virus utilizes its spike protein to attach to the human angiotensin-converting enzyme-2 (ACE2) receptor found on the cell surface [10]. Previously in SARS, the viruses predominantly infected the type-I alveolar (AT1) cells, although the majority (83%) of ACE2 is expressed in type-II alveolar (AT2) cells – indicating that AT2 cells could act as a potential reservoir for the viruses [11].
The ACE2 receptor is a type-I transmembrane protein with an enzymatically active domain expressed on the surface of many tissues such as lungs, arteries, heart, kidney, and intestine [9]. The primary function of hACE2 is to control vasoconstriction and regulate blood pressure by converting angiotensin-II into vasodilator angiotensin (1 to 7) [12]. As for SARS-CoV-2, the interaction with hACE2 remains to be the primary mechanism of entry into cells although there is a report of using basigin (CD147), also known as extracellular matrix metalloproteinase inducer (EMMPRIN), as a novel route of entry into cells [13].
The virus enters the cell via receptor-mediated endocytosis pathway [10]. The spike (S) protein is of central importance as it interacts with the host receptor and establishes the gateway for viral entry into the cell. The S-glycoprotein is a homotrimer and it has two halves, S1 and S2. The S protein is cleaved at the S1/S2 junction through transmembrane serine protease TMPRSS2; S1 binds to the receptor while S2 mediates fusion with the cell membrane for entry [14]. Inside the endolysosome, PIKfyve, TCP2, and Cathepsin L contribute to the viral entry. It was also shown that the virus could form syncytium independent of exogenous protease facilitating cell-to-cell fusion [10].
Following entry, the virus directs its genome translation by way of host ribosomes to produce polyproteins from ORF1a and ORF1b. The viral encoded proteases, 3-chymotrypsin-like protease (3CLpro) and papain-like protease (PLpro), cleave the polyproteins [15]. A multiprotein replicase-transcriptase complex (RTC) is formed from the non-structural proteins where RdRp plays the key role to produce genomic RNAs [16]. After generating a massive amount of copies, the viruses break free the cell’s borderline by activating different cell death programs. The SARS-CoV-2 exerts similar characteristics as of SARS-CoV in destroying the cells as evidenced by its lethality in cultured human cells [17]. Previously, autopsy studies from SARS showed that apoptosis was present in many organs, including the liver and thyroid gland [18]. In sputum sample, the average viral load was recorded as 7.00 × 106 RNA copies per ml with up to 2.35 × 109 copies per ml at maximum. The viral load was found to be the highest on day 4 with a progressive decline, and remained undetectable after day 28 [19].
DYSREGULATION OF RAAS AND KININ-KALLIKREIN SYSTEM
Experimental studies show that SARS-CoV-2 infection effectively downregulates the expression of ACE2 [20]. Since ACE2 is a key regulator of the RAAS (renin-angiotensin-aldosterone system) pathway, the downregulation of ACE2 can disrupt the balance between ACE/ACE2 and Ang-II/angiotensin-(1–7) [21]. An increased concentration of Ang-II was shown to aggravate lung injury through vascular permeability and pulmonary edema [22,23]. Of note, the RAAS consists of a network of regulatory proteins that operate in important processes, for instance, electrolyte balance, vascular permeability, and blood pressure regulation [21]. The RAAS pathway elevates blood pressure as renin initiates the system by converting angiotensinogen (produced by the liver) into angiotensin-I, and subsequently to angiotensin-II by ACE, acting as a positive regulator of the system to stimulate vasoconstriction. Conversely, the destruction of alveolar cells and downregulation of ACE2 cause overactivation of the RAAS system. The upregulation of Ang-II levels leads to the loss of the protective effects of angiotensin-(1–7) and contributes to severe lung injury, and the resulting damage releases inflammatory mediators leading to pyroptosis [18,24,25].
Moreover, the downregulation of ACE2 expression is hypothesized to have impacted the kinin-kallikrein system. The kallikreins are serine-protease enzymes that convert kininogen into kinin plasma protein. The activation of the kinin-kallikrein system releases bradykinins that bind to its receptor-B2 found on the endothelial cells to initiate relaxation of vascular smooth muscle and increase in vascular permeability [26]. Generally, the ACE and ACE2 work as kininase II enzymes to breakdown bradykinin metabolites; however, the downregulation of ACE2 impairs the balance in the system and increases bradykinin levels. Failure to neutralize the kinins can result in pulmonary injury, and angioedema [27].
INFLAMMASOME AND INFLAMMATORY MEDIATORS
The SARS-CoV-2 genome contains the viroporin 3a which is a potent activator of cytoplasmic nod-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome as seen in SARS-CoV [28]. A study suggests that proinflammatory cytokines are produced in response to the formation of NLRP3 inflammasome [28]. The inflammasomes are multiprotein complex in the cytosol that promote the secretion of IL-1β, IL-18, and damage-associated molecular patterns (DAMPs). Excessive release of DAMPs due to hyperactivation of NLRP3 inflammasome can result in the release of high mobility group box 1 (HMGB1), infiltration of neutrophils, activation of macrophages, pyroptosis, and cytokine production [29].
Studies show that a significantly elevated level of pro-inflammatory cytokines including IL-2, IL-7, IL-10, granulocyte-macrophage colony-stimulating factor (GM-CSF), interferon gamma-induced protein 10 (IP10), monocyte chemoattractant protein 1 (MCP1), macrophage inflammatory protein 1-alpha (MIP1α), and TNFα was found in patients affected with COVID-19 [30]. The activated CD4+ T cells, macrophages, dendritic cells, and epithelial cells secrete these cytokines and chemokines in response to inflammation [31–33]. The release of inflammatory mediators brings CD14+ CD16+ monocytes into pulmonary circulation where monocytes mature into macrophages and subsequently release notably IL-1. IL-6, TNFα and HMGB1 [8,34,35]. The HMGB1 acts both as a cytokine and a nuclear protein; and plays an important role in endothelial activation, and systemic inflammation [35]. The inflammation recruits natural killer (NK) cells and T cells producing GM-CSF, and tumor necrosis factor (TNF) inviting more macrophages to the site of infection [30].
The elevated levels of cytokines cause vasodilation and increase the capillary permeability by relaxing the smooth muscle and contracting the endothelial cells. This, in turn, allows blood plasma to leak into interstitial space, and to alveoli to cause alveolar edema. The pyrogenic cytokines travel via the blood to the central nervous system and get the hypothalamus to reset its thermostat to increase the body temperature. The inflammatory mediators, in particular IL-8, bring neutrophil to the scene to destroy the viruses by releasing reactive oxygen species (ROS) and an arsenal of enzymes into the surrounding tissues [36].
COLLATERAL DAMAGE: NEUTROPHILS AND HYPERACTIVE T CELLS
In COVID-19, laboratory tests found leucopenia with leukocyte counts of 2.91 × 109 cells/L of which 70.0% were neutrophils [30]. Neutrophils are important in viral infection as they keep the inflammation in check. Albeit, unless a balance is maintained between its beneficial and detrimental effects, they harm the host tissue by collateral damage [34]. The autopsy results from people with COVID-19 showed an extensive accumulation of neutrophil in pulmonary capillaries [37].
The damage of type-I and type-II pneumocytes impairs the gas exchange and surfactant production which inevitably leads to the increase of surface tension. The alveoli collapse and fluid containing proteins build up inside the alveoli. The cellular debris and consolidated proteinaceous fluid stimulate coughing, while the decrease in gas exchange results in hypoxia. An evaluation of the host immune system in COVID-19 critical patients found that the extent of hypoxemia was closely related to the levels of host immune cells [38]. The low partial pressure of oxygen prompts chemoreceptors to stimulate the sympathetic nervous system (SNS) to increase both heart rate and respiration rate; consequently, shortness of breath occurs.
The progression of the disease is also accounted for the reduced lymphocyte counts and the persistently low level of type-I interferon (IFN) induction [34]. Generally, the type-I IFN plays a crucial role in antiviral response by preventing viral replication and spread. However, it has been shown that beta coronaviruses can sequester their viral RNA from pattern recognition receptors (PRR) while the nonstructural proteins such as nsp3 and nsp1 can block the interferons to bind to its IFN receptors [39,40]. It is speculated that the SARS-CoV-2 infection might not effectively activate IFN-regulatory factor 3 (IRF3) and IRF7 which could contribute to reduced interferon response [39,41].
The global T cell lymphopenia seen in COVID-19 severe patients is yet to be clarified in detail, but tentative studies suggest that Fas-mediated activation-induced cell death (AICD) is the likely cause of lymphocytopenia and impaired immune response [42]. Moreover, the hyperactivated CD8 T cells with a high proportion of HLA-DR (CD4 3.47%) and CD38 (CD8 39.4%) were observed in COVID-19 [43]. In CD4 T cells, there was an increased concentration of highly proinflammatory CCR6+ Th17. The cytotoxic T lymphocytes (CTL) were equipped with high concentrations of cytotoxic granules: granzyme A, granzyme B, granzyme K, granzyme M and perforin [44]. The CTLs are supposed to initiate programmed cell death to prevent the survival of the invading virus but hyperreactive CTLs could show high cytotoxicity to induce apoptosis in healthy cells and lead to the subsequent severity of the disease [43].
THE COMPLEMENT CASCADES
As the first line of defense, the complement system exerts substantial action against various viral attacks [45]. However, in the case of SARS-CoV, one experimental study indicated that instead of protective effects, the complement system could potentially increase the chance for lung damage [46]. The C3a and C5a can recruit inflammatory cells to the site, and the recruitment of large amounts of neutrophil and macrophages can exert pro-inflammatory activity [47]. One study demonstrated that patients who died of ARDS in COVID-19 had an increased amount of CD163+ macrophages infiltration in the lungs and the macrophages expressed complement component 5a receptor 1 (C5aR1). The study also reported that the soluble level of C5a anaphylatoxin was linked to COVID-19 disease severity [48]. Another study reported that patients who suffered from respiratory failure in COVID-19 had an excessive amount of C5b-9, C4d, and MASP-2 deposition in the microvasculature [49]. Besides, the inflammatory chemokines such as CCL4 (macrophage inflammatory protein-1β), CCL2 (monocyte chemoattractant protein 1) and CXCL9 (monokine induced by gamma interferon) might have important roles in disease severity in COVID-19 [48].
IRON DYSREGULATION, GLUTATHIONE, AND FERROPTOSIS
Hyperferritinemia has been observed in COVID-19 patients and described as a marker of disease severity [50–52]. Iron plays an important role in oxygen transport from the lungs to the body; and the cells store irons as ferritin in its ferric (Fe3+) state. During infection, the interleukin-6 stimulates the synthesis of hepcidin which is a key regulator-hormone of iron, and hepcidin increases the intracellular concentration of ferritin [53,54]. The excess amount of intracellular irons interacts with molecular oxygen and generates ROS which can have damaging effects on the cells [55]. The accumulation of abundant accessible intracellular iron contributes to ferroptosis which is an iron-dependent regulated form of oxidative cell death [56].
Inside the cell, the glutathione (GHS) acts as an antioxidant and provides the primary protection against ferroptosis. Depletion of glutathione leads to loss of cellular antioxidant ability, and inhibition of enzymes dependent on glutathione, such as glutathione peroxidases [57]. One study showed that endogenous deficiency of glutathione can result in severe clinical complications in COVID-19 patients [58]. The glutathione deficiency was found to be associated with male sex, age over 65 years, chronic disease, cigarette smoking, and low glutathione intake linked to insufficient consumption of fresh vegetables [58]. One experimental study denoted that GSH deficiency and its related elevated oxidative stress could epigenetically change the expression of regulating genes of vitamin D contributing to a secondary vitamin D deficiency [59]. In summary, the disruption of iron homeostasis and the resulting ferroptosis can affect multiple organ systems including liver, kidney, heart, and lung [60].
EXTRAPULMONARY SYMPTOMS
A wide variety of clinical manifestations is observed in COVID-19 as the virus infects gastrointestinal, neurologic, renal, and myocardial tissues [8]. The gastrointestinal complications including anorexia, nausea, diarrhea, and abdominal pain could be directly attributed to viral invasion as SARS-CoV-2 was found to productively infect gut enterocytes [61]. Similarly, the manifestations of anosmia, hypogeusia, headache, and visual dysfunction suggest a potential retrograde axonal transport of the virus into the brain or via nasal mucosa, lamina cribrosa and the olfactory nerve [8]. Another study speculated the transport of the virus using motor proteins dynein and kinesin via afferent nerve endings from the lungs [62]. However, one study hypothesized that the disrupted sense of smell could ironically be an evolutionary mechanism to self-destruct olfactory neurons to halt viral tropism into the central nervous system [63]. Since the olfactory neurons regenerate every 30 – 120 days, the resulting anosmia could act as a self-defense mechanism which is supported by studies that indicate: anosmia may be a predictor of good prognosis for COVID-19 [64,65]. Despite, the evidence for viral tropism in neuron cells suggests that circulating leukocytes could alternatively transport SARS-CoV-2 across the blood-brain barrier in a “Trojan horse” manner [66].
High expression of ACE2 receptors in myocardial tissue and subsequent isolation of virus from these tissues support the possible mechanism of virus-mediated cardiac dysfunction, resulting in myocardial injury, arrhythmias, cardiomyopathy, and acute coronary syndromes (ACS) [8]. In severe patients, the low partial pressure of oxygen and hyper-inflammation led to the alteration of liver biomarkers and liver damage [67]. The elevated levels of cytokines may lead to apoptosis of pancreatic β-cell and a range of abnormalities such as worsened hyperglycemia and ketoacidosis. Endocrinologic complications were more pronounced in patients with pre-existing diabetes and obesity [8]. In addition, histopathologic studies suggest that the virus could directly infect the renal system damaging the kidney tubule cells; and in combination with hyperimmune reaction, the acute kidney injury (AKI) manifests as it impairs the normal body fluid flow [8]. The health records of 5,449 COVID-19 patients hospitalized in New York city hospitals revealed that up to 36.6% of them developed AKI, in which more than 30% were at stage 3, and often requiring renal replacement therapy (RRT) [68].
CYTOKINE STORM AND SYSTEMIC INFLAMMATION
In the cytokine storm, the elevated levels of cytokines contribute to vasodilation, systemic inflammation, and immune suppression. In the lungs, the widespread inflammation appears as acute respiratory distress syndrome (ARDS) and in conjunction with cytokines, it may cause systemic inflammatory response for some patients [69]. Respiratory failure could occur due to ARDS which is the likely cause of death in 70% of fatal COVID-19 cases [70]. Aging was found to be correlated with elevated proinflammatory cytokines which might imply why children are less affected in COVID-19 [71].
The endothelial activation due to the cytokine-release by activated neutrophils and macrophages secrete TNF-α and IL-1β. The formation of neutrophil extracellular traps (NETs) further damages the endothelium and stimulates the release of IL-6, IL-8, nitric oxide (NO), ROS, and platelet-activating factor (PAF) [8,34]. The subsequent vasodilation, increased permeability and decreased perfusion within the systemic circulation allow fluids to leak out and accumulate in the tissues. Blood volume and total peripheral resistance (TFR) decrease – resulting in the fall of blood pressure. With severe hypovolemia and low perfusion, hypovolemic shock takes place.
The systemic effects of cytokine storms recruit secondary immune mediators such as human cytokine synthesis inhibitory factor (CSIF) or IL-10 that suppresses cytokine production; and soluble tumor necrosis factor receptors (sTNFR) that neutralize circulating TNF-α and induce apoptosis. However, in COVID-19, all three types of immune responses (type-I, II, and III) are triggered and they subdue the buffering capacity of the host to maintain immune balance [72]. In response to high viral load, the IFN-α, IFN-γ, and TNF-α cytokines are overproduced and inflammasome-induced cytokines (i.e. IL-1β and IL-18) rise significantly [72]. One study demonstrated four distinct immune signatures where patients with severe outcomes had shown a sustained elevated cytokine response of all four types, particularly IFN-λ, TGF-α, IL-16, IL-23, IL-33 and coagulopathy markers [72].
MULTIPLE ORGAN DYSFUNCTION SYNDROME
The inflammation of the endothelium marked by the presence of activated neutrophils and macrophages, and the decreased expression of ACE2 after SARS-CoV-2 infection initiate pro‐inflammatory and procoagulant pathways [73]. The higher expression of tissue factor (TF) and plasminogen activator inhibitor 1 (PAI-1) and the impairment of tissue factor pathway inhibitor (TFPI) allow the formation of fibrin-based blood clots throughout the bloodstream, particularly in the capillaries [34]. One study provided biochemical evidence and showed that endotheliopathy was widespread among critically ill patients of COVID-19 [74].
The occurrence of thromboinflammation causes disseminated intravascular coagulation (DIC) leading to fibrin deposition and microvascular thrombosis. Study shows that hypoxia-mediated hyperviscosity and the upregulation of the HIF-1 (hypoxia-inducible factor 1) may contribute prothrombotic state [75]. The deposition of microthrombi and vasculitis could manifest as cutaneous manifestation. The thrombi could travel through the circulatory system and create a blockage in the supply of blood and oxygen in a particular organ, resulting in tissue ischemia. The occurrence of pulmonary embolism and deep venous thrombosis (DVT) has been frequently reported in COVID-19 pneumonia patients [76]. Moreover, emerging evidence suggests that thrombosis could take place in intravenous catheters, extracorporeal circuits leading to myocardial infarction, and ischemic stroke [8]. Serum inflammatory markers such as C-reactive protein (CRP), D-dimer, and fibrinogen are prognostic of subsequent severity and mortality in COVID-19 [8,77,78].
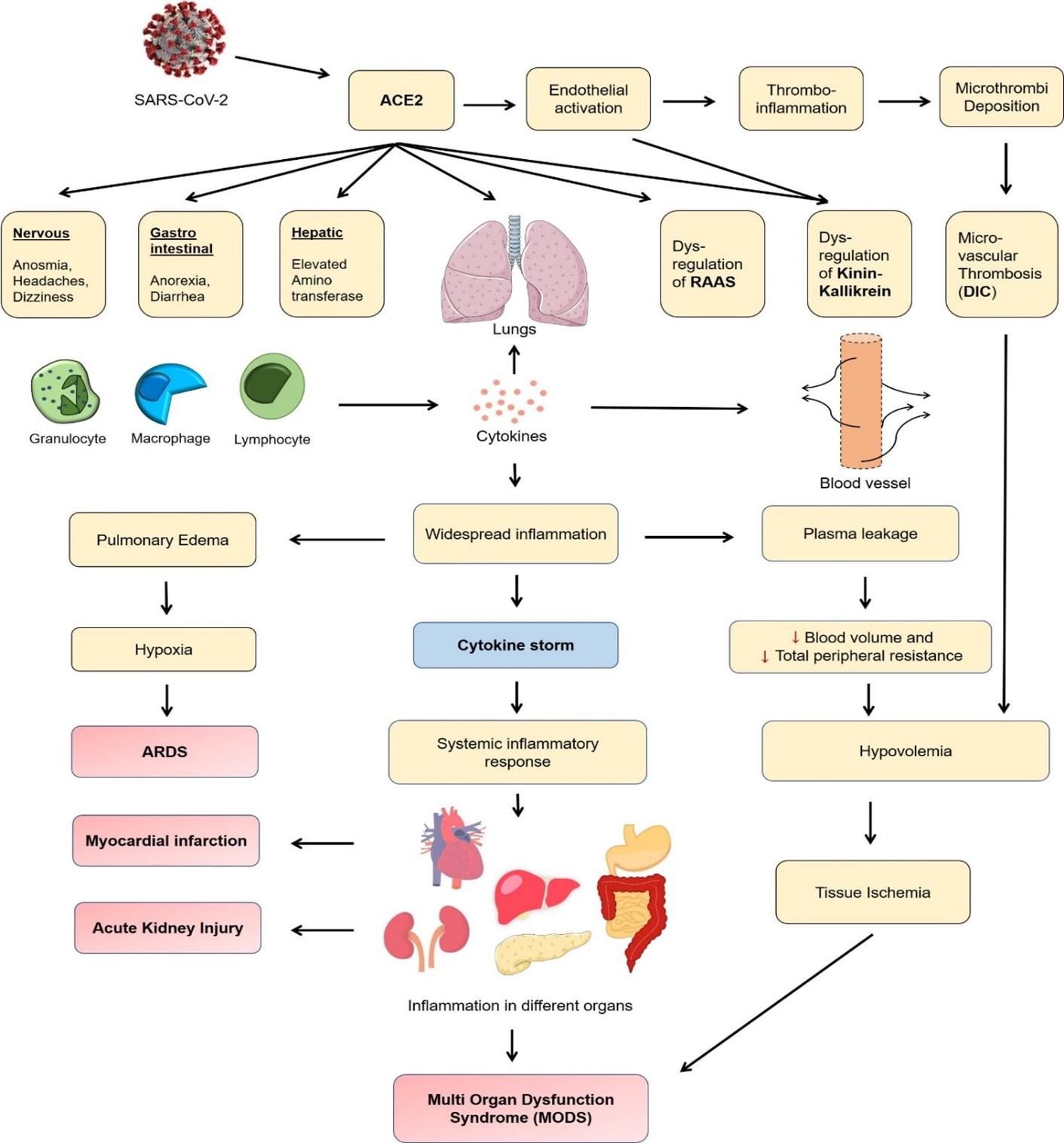
With cytokine storm mediated systemic inflammation, hypoxia, hypotension, and thrombosis, the multi-organ damage takes place especially in the form of cardiac, hepatic, and renal failure, leading to multi-organ dysfunction syndrome (MODS). The physiologic derangement associated with MODS can involve two or more organ systems failure, eventually, progress to an irreversible state and death. Figure 1 shows a schematic diagram of COVID-19 pathophysiology leading to multi organ failure.
FUTURE PERSPECTIVES
With the ability to invade multiple organ systems and disrupt a wide range of pathways, the COVID-19 has presented itself as a complex biological phenomenon that remains to be elucidated in detail. Areas of major interests include the proper understanding of how the downregulation of ACE2 affects the kinin-kallikrein system (KKS) and blood pressure; and how the development of thrombotic complications in COVID-19 is associated with the kinin-kallikrein system.
Since type-I interferons are part of the body’s immune-combat strategy against viruses, the cause for the reduced type-I interferon production in COVID-19 patients is an open area of research. In addition, the evidence for the direct invasion of SARS-CoV-2 in different cell types will uncover the degree and extent of infection. T cell lymphopenia, iron dysregulation, and neural tropism are also among the highlights for further research.
It is notable to mention that multiple organ dysfunction syndrome (MODS) is suggested to be different from the multisystem inflammatory syndrome in children (MIS-C) [79]. The MIS-C is likely the result of an autoimmune response mediated by antibodies affecting a number of organs; and the syndrome may lead to fatality. The potential for antibody-dependent enhancement (ADE) needs to be investigated since ADE may exert an adverse outcome in vaccination. With an in-depth understanding of pathogenesis and pathophysiology, it will be possible to devise effective strategies to fight the COVID-19.
CONCLUSIONS
The COVID-19 pandemic has become a global concern as the disease is likely to persist for a long time. The wide range of clinical manifestations beyond the respiratory system warrants rigorous research to develop a comprehensive scenario of COVID-19 disease. topics including the mechanism of direct viral injury, the potential alternative routes of invasion, the role of bradykinins in blood clots, ferroptosis, T cell apoptosis, and nerve cell damage in anosmia require special focus to answer the points at issue. We believe our updated study of COVID-19 pathophysiology will contribute to a holistic understanding of the mechanism of disease development, and thus provide a window for better strategies in therapeutic interventions and patient care.
ACKNOWLEDGEMENT
This research received no external funding.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
MHB Siam designed the study and wrote the manuscript. MHB Siam, and SMM Abedin carried out the literature review. A Ahmed and MHB Siam performed the illustration. MS Hossain supervised the study. NH Nishat, MS Hossain, and A Ahmed provided critical intellectual contribution in reviewing and revising the manuscript. All authors read and approved the final submitted version of the manuscript.
References
- [1]Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med 2020;26:450 –2.
- [2]Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 2020;395:565–74.
- [3]Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA – J Am Med Assoc 2020;324:782–93.
- [4]Sudre CH, Lee K, Ni Lochlainn M, Varsavsky T, Murray B, Graham MS, et al. Symptom clusters in Covid19: A potential clinical prediction tool from the COVID Symptom study app. MedRxiv 2020:2020.06.12.20129056.
- [5]Chen J, Lau YF, Lamirande EW, Paddock CD, Bartlett JH, Zaki SR, et al. Cellular Immune Responses to Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) Infection in Senescent BALB/c Mice: CD4+ T Cells Are Important in Control of SARS-CoV Infection. J Virol 2010;84:1289–301.
- [6]COVID-19 Pandemic Planning Scenarios | CDC n.d. https://www.cdc.gov/coronavirus/2019-ncov/hcp/planning-scenarios.html (accessed August 4, 2020).
- [7]De Wit E, Van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: Recent insights into emerging coronaviruses. Nat Rev Microbiol 2016;14:523–34.
- [8]Gupta A, Madhavan M V., Sehgal K, Nair N, Mahajan S, Sehrawat TS, et al. Extrapulmonary manifestations of COVID-19. Nat Med 2020;26.
- [9]Hamming I, Timens W, Bulthuis MLC, Lely AT, Navis GJ, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 2004;203:631–7.
- [10]Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun 2020;11:1–12.
- [11]hao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W. Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. BioRxiv 2020:2020.01.26.919985.
- [12]Keidar S, Kaplan M, Gamliel-Lazarovich A. ACE2 of the heart: From angiotensin I to angiotensin (1-7). Cardiovasc Res 2007;73:463–9.
- [13]Wang K, Chen W, Zhou Y-S, Lian J-Q, Zhang Z, Du P, et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. BioRxiv 2020:2020.03.14.988345.
- [14]Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020;181:271-280.e8.
- [15]Chen YW, Yiu CPB, Wong KY. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure: Virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research 2020;9.
- [16]Fehr AR, Perlman S. Coronaviruses: An overview of their replication and pathogenesis. Coronaviruses Methods Protoc., vol. 1282, Springer New York; 2015, p. 1–23.
- [17]Zhou P, Yang X Lou, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020;579:270–3.
- [18]Chau TN, Lee KC, Yao H, Tsang TY, Chow TC, Yeung YC, et al. SARS-Associated Viral Hepatitis Caused by a Novel Coronavirus: Report of Three Cases. Hepatology 2004;39:302–10.
- [19]Wölfel R, Corman VM, Guggemos W, Seilmaier M, Zange S, Müller MA, et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020;581:465–9.
- [20]Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med 2020;46:586–90.
- [21]Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005;436:112–6.
- [22]Marshall RP, Gohlke P, Chambers RC, Howell DC, Bottoms SE, Unger T, et al. Angiotensin II and the fibroproliferative response to acute lung injury. Am J Physiol – Lung Cell Mol Physiol 2004;286:156–64.
- [23]Tan WSD, Liao W, Zhou S, Mei D, Wong WSF. Targeting the renin–angiotensin system as novel therapeutic strategy for pulmonary diseases. Curr Opin Pharmacol 2018;40:9–17.
- [24]Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med 2005;11:875–9.
- [25]Ni W, Yang X, Yang D, Bao J, Li R, Xiao Y, et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit Care 2020;24:422.
- [26]Veerdonk F van de, Netea MG, Deuren M van, Meer JWM van der, Mast Q de, Bruggemann RJ, et al. Kinins and Cytokines in COVID-19: A Comprehensive Pathophysiological Approach. Preprints 2020:1–29.
- [27]Cohen AJ, DiFrancesco MF, Solomon SD, Vaduganathan M. Angioedema in COVID-19. Eur Heart J 2020.
- [28]Chen I-Y, Moriyama M, Chang M-F, Ichinohe T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front Microbiol 2019;10:50.
- [29]van den Berg DF, te Velde AA. Severe COVID-19: NLRP3 Inflammasome Dysregulated. Front Immunol 2020;11:1580.
- [30]Rothan HA, Byrareddy SN. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J Autoimmun 2020;109.
- [31]Coperchini F, Chiovato L, Croce L, Magri F, Rotondi M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev 2020;53:25–32.
- [32]Maurer M, Von Stebut E. Macrophage inflammatory protein-1. Int J Biochem Cell Biol 2004;36:1882–6.
- [33]azzucchelli RI, Warming S, Lawrence SM, Ishii M, Abshari M, Washington AV, et al. Visualization and Identification of IL-7 Producing Cells in Reporter Mice. PLoS One 2009;4:e7637.
- [34]Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol 2020;20:355–62.
- [35]Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat Rev Immunol 2005;5:331–42.
- [36]Bardoel BW, Kenny EF, Sollberger G, Zychlinsky A. The balancing act of neutrophils. Cell Host Microbe 2014;15:526–36.
- [37]Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020;5.
- [38]Zhou G, Chen S, Chen Z. Advances in COVID-19: the virus, the pathogenesis, and evidence-based control and therapeutic strategies. Front Med 2020;14:117–25.
- [39]Acharya D, Liu GQ, Gack MU. Dysregulation of type I interferon responses in COVID-19. Nat Rev Immunol 2020;20:397–8.
- [40]Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016;19:181–93.
- [41]Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020;181:1036-1045.e9.
- [42]chen yongwen, Feng Z, Diao B, Wang R, Wang G, Wang C, et al. The Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Directly Decimates Human Spleens and Lymph Nodes. MedRxiv 2020:2020.03.27.20045427.
- [43]Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med 2020;8:420–2.
- [44]Thevarajan I, Nguyen THO, Koutsakos M, Druce J, Caly L, van de Sandt CE, et al. Breadth of concomitant immune responses prior to patient recovery: a case report of non-severe COVID-19. Nat Med 2020;26:453–5.
- [45]Agrawal P, Nawadkar R, Ojha H, Kumar J, Sahu A. Complement evasion strategies of viruses: An overview. Front Microbiol 2017;8:1117.
- [46]Maglakelidze N, Manto KM, Craig TJ. A Review: Does Complement or the Contact System Have a Role in Protection or Pathogenesis of COVID-19? Pulm Ther 2020:1–8.
- [47]Abou-El-Hassan H, Zaraket H. Viral-derived complement inhibitors: current status and potential role in immunomodulation. Exp Biol Med 2017;242:397–410.
- [48]Carvelli J, Demaria O, Vély F, Batista L, Benmansour NC, Fares J, et al. Association of COVID-19 inflammation with activation of the C5a-C5aR1 axis. Nature 2020.
- [49]Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res 2020;220:1–13.
- [50]Edeas M, Saleh J, Peyssonnaux C. Iron: Innocent bystander or vicious culprit in COVID-19 pathogenesis? Int J Infect Dis 2020;97:303–5.
- [51]Phua J, Weng L, Ling L, Egi M, Lim CM, Divatia JV, et al. Intensive care management of coronavirus disease 2019 (COVID-19): challenges and recommendations. Lancet Respir Med 2020;8:506–17.
- [52]Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 2020;395:1033–4.
- [53]McDermid JM, Hennig BJ, van der Sande M, Hill AVS, Whittle HC, Jaye A, et al. Host iron redistribution as a risk factor for incident tuberculosis in HIV infection: An 11-year retrospective cohort study. BMC Infect Dis 2013;13:1–9.
- [54]Daher R, Manceau H, Karim Z. Iron metabolism and the role of the iron-regulating hormone hepcidin in health and disease. Press Medicale 2017;46:e272–8.
- [55]Kell DB, Pretorius E. Serum ferritin is an important inflammatory disease marker, as it is mainly a leakage product from damaged cells. Metallomics 2014;6:748–73.
- [56]Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic Biol Med 2020;152:175–85.
- [57]Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol 2016;26:165–76.
- [58]Polonikov A. Endogenous Deficiency of Glutathione as the Most Likely Cause of Serious Manifestations and Death in COVID-19 Patients. ACS Infect Dis 2020.
- [59]Parsanathan R, Jain SK. Glutathione deficiency induces epigenetic alterations of vitamin D metabolism genes in the livers of high-fat diet-fed obese mice. Sci Rep 2019;9:1–11.
- [60]Sun Y, Chen P, Zhai B, Zhang M, Xiang Y, Fang J, et al. The emerging role of ferroptosis in inflammation. Biomed Pharmacother 2020;127:110108.
- [61]Lamers MM, Beumer J, van der Vaart J, Knoops K, Puschhof J, Breugem TI, et al. SARS-CoV-2 productively infects human gut enterocytes. Science 2020;369:50–4.
- [62]Ahmed MU, Hanif M, Ali MJ, Haider MA, Kherani D, Memon GM, et al. Neurological Manifestations of COVID-19 (SARS-CoV-2): A Review. Front Neurol 2020;11:518.
- [63]Le Bon SD, Horoi M. Is anosmia the price to pay in an immune-induced scorched-earth policy against COVID-19? Med Hypotheses 2020;143:109881.
- [64]Oboti L, Peretto P, de Marchis S, Fasolo A. From chemical neuroanatomy to an understanding of the olfactory system. Eur J Histochem 2011;55:194–9.
- [65]Romero-Sánchez CM, Díaz-Maroto I, Fernández-Díaz E, Sánchez-Larsen Á, Layos-Romero A, García-García J, et al. Neurologic manifestations in hospitalized patients with COVID-19: The ALBACOVID registry. Neurology 2020:10.1212/WNL.0000000000009937.
- [66]Huang J, Zheng M, Tang X, Chen Y, Tong A, Zhou L. Potential of SARS-CoV-2 to Cause CNS Infection: Biologic Fundamental and Clinical Experience. Front Neurol 2020;11:659.
- [67]Gholizadeh P, Safari R, Marofi P, Zeinalzadeh E, Pagliano P, Ganbarov K, et al. Alteration of Liver Biomarkers in Patients with SARS-CoV-2 (COVID-19). J Inflamm Res 2020;Volume 13:285–92.
- [68]Hirsch JS, Ng JH, Ross DW, Sharma P, Shah HH, Barnett RL, et al. Acute kidney injury in patients hospitalized with COVID-19. Kidney Int 2020;98:209–18.
- [69]Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the Eye of the Cytokine Storm. Microbiol Mol Biol Rev 2012;76:16–32.
- [70]Zhang B, Zhou X, Qiu Y, Feng F, Feng J, Jia Y, et al. Clinical characteristics of 82 death cases with COVID-19. MedRxiv 2020:2020.02.26.20028191.
- [71]Yuki K, Fujiogi M, Koutsogiannaki S. COVID-19 pathophysiology: A review. Clin Immunol 2020;215:108427.
- [72]Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020:1–9.
- [73]Bourgonje AR, Abdulle AE, Timens W, Hillebrands J, Navis GJ, Gordijn SJ, et al. Angiotensin‐converting enzyme 2 (ACE2), SARS‐CoV‐2 and the pathophysiology of coronavirus disease 2019 (COVID‐19). J Pathol 2020;251:228–48.
- [74]Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol 2020;7:e575–82.
- [75]Gupta N, Zhao YY, Evans CE. The stimulation of thrombosis by hypoxia. Thromb Res 2019;181:77–83.
- [76]Bompard F, Monnier H, Saab I, Tordjman M, Abdoul H, Fournier L, et al. Pulmonary embolism in patients with Covid-19 pneumonia. Eur Respir J 2020:2001365.
- [77]Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern Med 2020;180:934.
- [78]Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 2020;395:1054–62.
- [79]Jiang L, Tang K, Levin M, Irfan O, Morris SK, Wilson K, et al. COVID-19 and multisystem inflammatory syndrome in children and adolescents. Lancet Infect Dis 2020;0.