Prediction of SARS-CoV-2 main protease inhibitors in medicinal plant-derived compounds by molecular docking approach
Abstract
Coronaviruses are endemic in humans and infections typically mild, such as the common cold. Still, the cross-species transmission has produced some unusually virulent strains which now causing viral pneumonia, in severe cases, even acute respiratory distress syndrome and death. SARS-CoV-2 is the most threatening issue which leads the world to an uncertainty alongside thousands of regular death scenes. An effective vaccine to cure this virus is not yet available, so it requires concerted efforts at various scales. The viral Main Protease controls coronavirus replication and is a proven drug discovery target for SARS-CoV-2. Comprehensive computational study e.g., molecular docking and ADMET (absorption, distribution, metabolism and excretion) profiling were employed to predict the efficacy of medicinal plant-based bioactive compounds against SARS-CoV-2 MPP. Paritaprevir and lopinavir-previously approved viral main protease inhibitors were used as standards for comparison. MPP was docked with 90 phytochemical compounds, and the screening revealed that four compounds (azadirachtin, -12.5 kcal/mol; rutin, -9 kcal/mol; theaflavin, -9 kcal/mol; astragalin, -8.8 kcal/mol) showed the highest binding affinity than the controls paritaprevir and lopinavir (-8.7 and -7.9 kcal/mol, respectively). Comparative structural analysis of protein-inhibitor complexes revealed that the compounds have intense interaction with the vital catalytic residue His-41 and Cys-145. Furthermore, the pharmaco-kinetics and drug-likeness properties of the antiviral phytochemicals suggested that the compounds do not have any considerable detrimental effects and can be considered potential drug candidates against SARS-CoV-2. These compounds can be further explored for in vitro experimental validation against SARS-CoV-2.
INTRODUCTION
The recently pandemic Corona Virus Disease 19 (COVID-19) has become a serious, rapidly growing global public health issue of an unprecedented level [1]. It is caused by prevalence of the infectious and pathogenic novel coronavirus named SARS-CoV-2 (Severe Acute Respiratory Syndrome-Coronavirus-2) [2]. As of October 9, SARS-CoV-2 infection has been reported in 216 countries, with 36.24 million confirmed cases and 1054868 total deaths [2, 3]. It can transmit from one individual to other by respiratory droplets. SARS-CoV-2 infected patients have general signs and symptoms, suffering initially from common flu-like fever, dry cough, dyspnoea, headache, sore throat, and diarrhea, which may further lead to express life-threatening symptoms including fatal pneumonia [4, 5].
Moreover, these outbreaks have affected the global economy, causing high economic losses including international trade and tourism [6]. The efficacy and safety of antivirals require evaluation by clinical trial. Currently, there is no efficient, safe, and specific potential drugs, vaccines to be approved for rapid remedy of this new respiratory syndrome [7, 8]. Hence, there is an urgent need to find new promising drug candidates to check and control the virus.
SARS-CoV-2 is a betacoronavirus similar to MERS-CoV and SARS-CoV, causing outbreaks with pandemic potential [9]. But they also showed dissimilarities that can influence their process of pathogenesis [10, 11]. These coronavirus genomes are enveloped, single-stranded positive-sense RNA of about 26-30 kb in size and consist of a minimum of six open reading frames (ORFs) that synthesis at least 4 structural and 16 nonstructural proteins [12,13]. ORF 1a/b is translated into a large protein that undergoes extensive proteolytic processing to produce the replicase complex, which mediates viral transcription and replication [12]. The protease responsible for the proteolytic processing is the main protease (MPP) or 3C-like protease (3CLpro), which is matured by auto-cleavage into the dimeric active conformation [14]. The crystallized form of SARS-CoV-2 main protease (MPP) was demonstrated by a Chinese researcher Liu et al. 2020 [15] that it is a potential drug target protein for the inhibition of SARS-CoV-2 replication. Thus, targeting MPP can provide effective treatment against SARS-CoV-2 by inhibition of the viral polypeptide cleavage.
Some preliminary experiments have been designed to find out effective combinations, including protease inhibitor lopinavir/ritonavir, which is commonly used to treat human immunodeficiency virus (HIV), for the medication of SARS-CoV-2 patients [16]. Other reported antiviral treatments form human pathogenic CoVs include nucleoside analogues, neuraminidase inhibitors, remdesivir, umifenovir, tenofovir disoproxil (TDF), and lamivudine (3TC) [17]. A separate research executed by Xu et al. 2020 implied that among 4 tested drugs (nelfinavir, pitavastatin, perampanel, and praziquantel), nelfinavir was identified as the best potential inhibitor against SARS-CoV-2 MPP, based on binding free energy calculations using the molecular mechanics with generalised Born and surface area salvation (MM/GBSA) model and solvated interaction energy (SIE) methods [18]. Many scientists reported the application of medicinal plants and their therapeutic uses as drugs from the ancient times [19]. Moreover, the expansion of natural products as new medicine or drug to resist the emerging virus SARS-CoV-2 could bypass the side effects of synthetic drugs. Therefore, our present study designed to screen out several antiviral plant-based compounds as potential inhibitor candidates for SARS-CoV-2 MPP through molecular docking approach.
MATERIALS AND METHODS
Phylogenetic and pairwise sequence alignment analysis
A multiple sequence and structure alignment analysis was performed to identify evolutionarily conserved functional residues among SARS-CoV-2, SARS-CoV and MERS-CoV that could be used as a target for the discovery of drug hits. Sequences of SARS-CoV-2 (PDB ID 6LU7) [20], SARS-CoV (PDB ID 2A5I) [21] and MERS-CoV (PDB ID 5WKK) [22] main proteases were retrieved from the Protein Data Bank (PDB) [23]. Retrieved protein sequences were allowed to multiple sequence alignment (MSA) by clustalW [24] and phylogenetic relationship (Neighbor-joining) study by using MEGA X [25] to understand the hereditary origin and pathogenicity of SARS-CoV-2 with other coronaviruses. Structural alignment/superposition analysis was also carried out to ensure broad-spectrum relevance of these protein targets using the Pymol version 1.7.4.5 Edu [26].
Retrieval of the structure and preparation of target
The 3D structure of the Main Protease Proteins (MPP) essential for replication of SARS-Cov-2 essential for virus replication (PDB ID: 6LU) [27, 20] was collected from RCSB Protein Data Bank [23], in .pdb format.
Before molecular docking, the 3D structure of MPP was processed by removing water molecules and ligand using Biovia Discovery Studio 4.5 [28] as it was in a complex structure with an inhibitor and energy was minimized by steepest descent and conjugate gradient techniques. The GROMACS 96 43B1 algorithm in SWISS-PDB viewer [29] and Chimera (Amber Force field) were conducted to prepare the final target receptor protein [30].
Ligand preparation
A comprehensive library of the medicinal plants containing phytochemicals with potential antiviral activity and traditional medicinal compounds was produced by searching in Dr. Duke’s phytochemical and ethnobotanical databases and by searching related literature in previously published studies (Supplementary Table 1) and screened against the SARS-CoV-2 MPP. The 3-dimensional (3D) structure of all compounds was obtained from PubChem, in .sdf format, and Open Babel software was used to convert SDF format compounds to PDB format [31]. For optimization and ligand preparation, we used PyRx [32] integrated mmff94 (Merck molecular force field) force field [33]. The ligands were then converted into PDBQT format.
Determination of active sites
The amino acids present in the active site of a protein were determined using the Computed Atlas for Surface Topography of Proteins (CASTp) and Biovia Discovery Studio 4.5 [28]. The determination of the amino acids in the active site was used to analyze the Grid box and docking evaluation results.
Molecular docking
The PyRx [32] software of the molecular docking approach was employed to screen the drugs against the Main Protease Protein of SARS-CoV-2. The protein was exposed to 90 phytochemicals for analyzing the highest negative binding energy and interactive amino acids. Recent drug repurposing studies proposed a few drugs that target SARS-CoV-2 MPP, suggesting them for the treatment of SARS-CoV-2. Herein, we selected the best of these (paritaprevir and lopinavir) from different drug repurposing studies [34] and docked them as controls in the present study. The grid box parameters were set to a size of 60 Å × 70 Å × 62 Å (x × y × z) and center of -10.5011 Å × 13.9110 Å × 67.9200 Å (x × y × z). LigPlot+ was used to generate the 2D ligand-protein interaction diagrams and determine the involved amino acids with their interactive position in the docked molecules [35]. Discovery Studio and Pymol version 1.7.4.5 Edu were used to visualize and analyze the ligand molecules’ interactions with the viral proteins [28, 26].
Drug likeness properties analysis of the screened compounds
The absorption, distribution, metabolism, and excretion (ADME) properties of the topmost phytochemical candidates for the MPP inhibitors were assessed by using the Swiss ADME portal [36]. In our study, the physico-chemical parameters (formula molecular weight, molar refractivity, TPSA), lipophilicity (Log Po/w (iLOGP), Log Po/w (XLOGP3), Log Po/w (WLOGP), Log Po/w (MLOGP), Log Po/w, (SILICOS-IT), Consensus Log Po/w), and water solubility (Log S: SILICOS-IT, solubility) of the topmost screened MPP inhibitors were checked out. Furthermore, selected MPP inhibitors have been used to test the inhibitory effects with various CYP isoforms (CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4). However, other relevant pharmacokinetic parameters, such as gastrointestinal (GI) absorption, BBB (blood-brain barrier) permeant, and P-gp substrate, have also been studied for potential drug candidates for main protease proteins. [37]
RESULTS
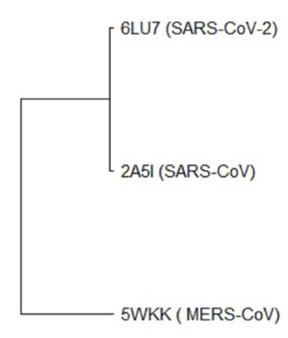
Phylogeny analysis and pairwise sequence alignment
The sequence alignment exhibited that the SARS-CoV-2 MPP is 96.08% and 47.71% identical to SARS-CoV (PDB: 2A5I) and MERS-CoV (PDB: 5WKK) main proteases, respectively. It had been found that SARS-CoV-2 was evolutionarily related to SARS-CoV (Figure 1) as SARS-CoV-2 aligned with the same clade of SARS-CoV where MERS-CoV was found in divergent relation with SARS-CoV-2. The sequence alignment also revealed that the catalytic dyad residues His41 and Cys145 of SARS-CoV-2 main protease are conserved among SARS-CoV-2, SARS-CoV, and MERS-CoV. Other residues of active sites are common in the main proteases of all 3 coronaviruses (Figure 2).
Screening of MPP inhibitors against the MPP of SARS-CoV-2
The main protease (MPP) or 3-chymotrypsin-like protease (3CLpro) or Nsp5 plays a vital role in cleaving the viral polyprotein at eleven different sites to form various Nsp required for viral replication [38]. Nsps maturation, which is necessary in the life cycle of the virus is mediated directly by MPP. Detailed study of the MPP catalytic mechanism makes it an attractive target for drug development against COVID-19 [39]. Information on the main protease protein is represented in Table 1.
Table 1. Protein target structures and active site amino acids (Biovia Discovery Studio 4.5, 2019) and the native ligand structure.
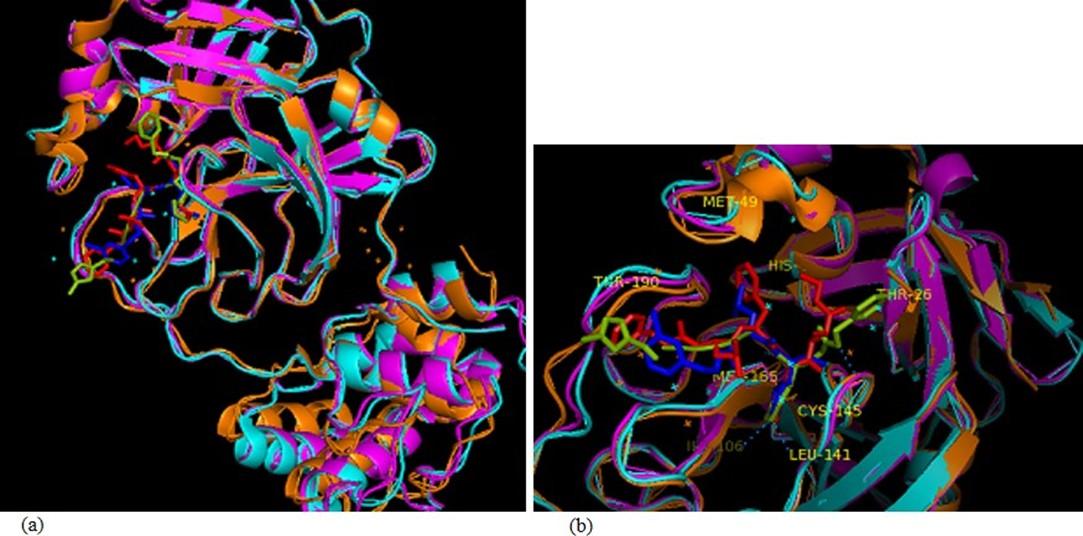
Two approved drugs for MPP inhibitors- paritaprevir [40] and lopinavir [41] were used as a positive control for the screening of antiviral phytochemicals as potential drugs. All the listed phytochemicals and MPP inhibitors were employed for molecular docking by using PyRx-virtual screening tool. Biovia Discovery Studio 4.5 was utilized to predict the interaction between the mentioned ligands and the MPP of SARS-CoV-2. List of phytochemicals that showed good results in docking with their medicinal activities are represented (Table 2) [42-59].Two approved drugs for MPP inhibitors- paritaprevir [40] and lopinavir [41] were used as a positive control for the screening of antiviral phytochemicals as potential drugs. All the listed phytochemicals and MPP inhibitors were employed for molecular docking by using PyRx-virtual screening tool. Biovia Discovery Studio 4.5 was utilized to predict the interaction between the mentioned ligands and the MPP of SARS-CoV-2. List of phytochemicals that showed good results in docking with their medicinal activities are represented (Table 2) [42-59].
The binding affinities obtained from the docking of MPP with all 90 selected ligands are represented in the table 3. Interaction and docking information of selected top 18 antiviral medicinal plant compounds with two established MPP inhibitors are enlisted in Table 4. Among the screened antiviral phytochemicals, azadirachtin- from medicinal plant Azadirachta indica showed the highest binding affinity -12.5 (kcal/mol), which was followed by rutin, extract from Nigella sativa represented -9.0 (kcal/mol) and theaflavin from Allium sativum -9.0 (kcal/mol). In comparison, two approved MPP inhibitors produced -8.2 and -7.9 (kcal/mol), respectively, in our study. Previous studies have demonstrated Cys-145 a key residue in the active site of SARS-CoV MPP, which makes it an important target for covalent inhibitors [60, 61, 62]. Moreover, the native ligand (N3) of our selected MPP also interacts with the catalytic dyad Cys145-His41 [63]. The docked compounds interaction indicates that all selected compounds interact with either catalytic residues His-41 and Cys-145 or at least one of them. It has been shown that an active site of main protease, where the three highest point compounds bind to incorporated hydrophobic residues such as Met-49, Leu-141, Cys-145, Met-165, Leu-167 and Pro-168, in addition to the polar contribution of amino acids such as Thr-25, Thr-26, His-41, Asn-142, Ser-144, His-163 and His-164. Hydrogen bonding with Leu-141 and Glu-166 also stabilized their conformations. Similar result was found in Kumar et al. 2020 [64] and da Silva Hage-Melim et al. 2020 [65]. Moreover, the ligand forms interaction with other substrate-binding pocket residues shown in figure 4 and table 4.
Here, Paritaprevir was found to be involved with the amino acid His-41, Met-49, Leu-141, Asn-142, Gly-143, Cys-145, His-164, Met-165, Glu-166, Leu-167, Pro-168, Asp-187, Arg-188, Gln-189, Thr-190, Gln-192 in the MPP of SARS-CoV-2 (Figure 3). Azadirachtin exhibited the highest binding energy at the active site of COVID-19 and it formed interactions with His-41,Ser-46 Met-49, Phe-140, Leu-141, Asn-142, Gly-143, Cys-145, His-163, His-164, Met-165, Glu-166, Pro-168, His-172, Gln-189, Thr-190, Ala-191 (Figure 4a, Table 4). Results of this study shown that Thr-26, Leu-27, His-41, Met-49, Pro-52, Tyr-54, Phe-140, Leu-141, Asn-142, Gly-143, Ser-144, Cys-145, His-163, His-164, Met-165, Glu-166, Leu-167, Pro-168, His-172, Asp-187, Arg-188, Gln-189 were critical residues for the binding of rutin to protease protein (Figure 4b, Table 4). Active site residues Leu-27, Ser- 46, Met-49, Phe-140, Leu-141, Asn-142, Gly-143, Ser-144, Cys-145, His-163, Mer-165, Glu-166, His-172, Arg-188, Gln-189, Thr-190, Gln-192 participated in interactions with theaflavin (Figure 4c, Table 4).
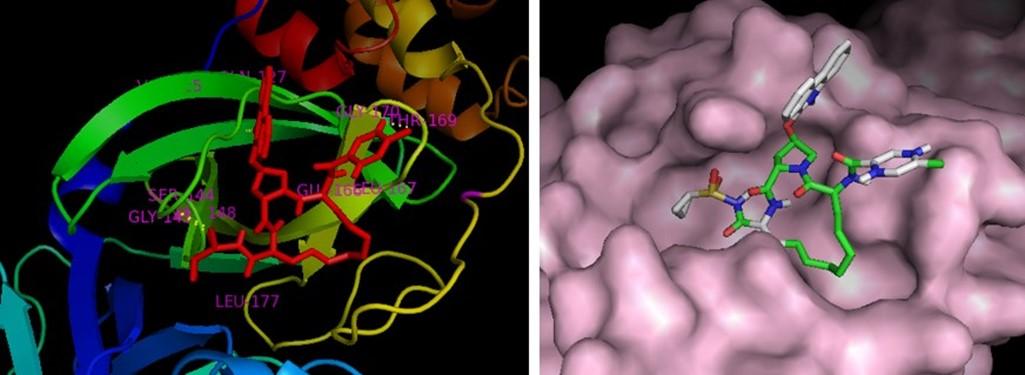
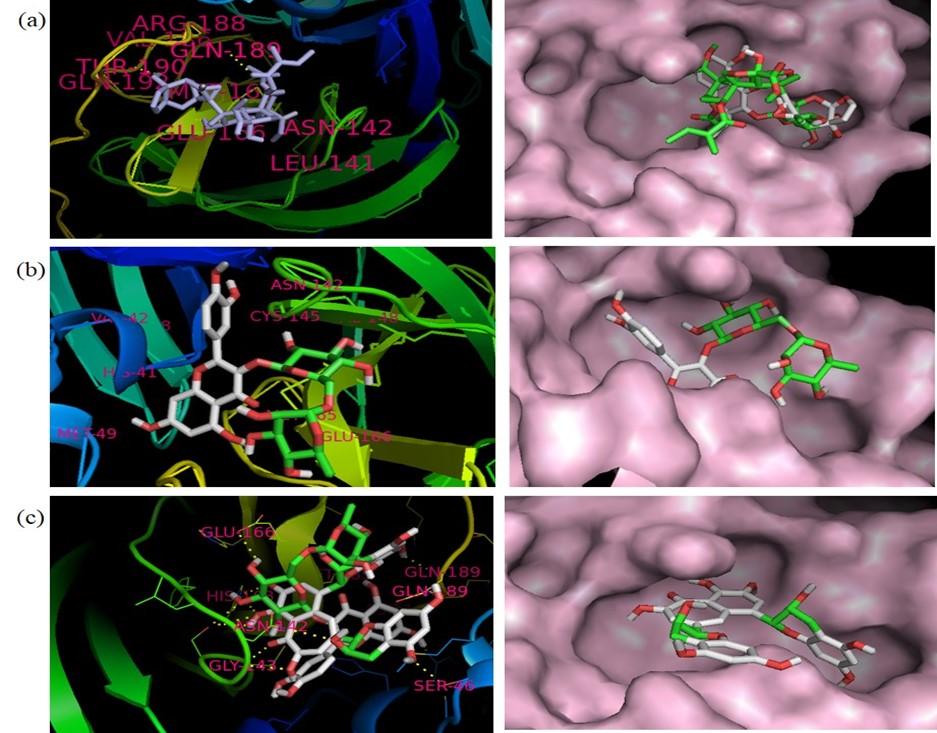
Table 2. List of top-ranked antiviral phytochemicals screened against SARS-CoV-2 MMP receptor binding site with respective sources and activities.
Table 3. Sources and Molecular Docking results of all the phytochemicals studied as antiviral agent.
Table 4. Molecular docking results with interactive amino acids from SARS-CoV-2 MPP of top phytochemicals and approved MPP inhibitors.
Drug likeness properties analysis of the screened phytochemicals
Different ADME properties, i.e., physicochemical parameters, pharmacokinetics, lipophilicity, water solubility, medicinal chemistry of top inhibitors were assumed to evaluate their pharmacological profile and described in Table 5. Analysis of the inhibitory effects with different CYP isoforms (CYP1A2, CYP2D6, CYP2C9, CYP2C19, CYP3A4) revealed that only theaflavin had an inhibitory effect on CYP2C9 and CYP3A4, while other MPP inhibitors had no interaction with the cytochromes P450 (CYP) isoforms. Remarkably, none of the screened compounds showed any undesired effects such as mutagenicity, tumorigenicity, irritating and reproductive effects. However, GI absorption was found low in case of every phytochemicals. The blood-brain barrier (BBB) permeation was also calculated by BOILED-Egg models [66] and among the putative MPP inhibitors, there was no BBB permeability found. Saponins and aloin show the highest solubility, while each candidate is soluble in water to moderate to high levels.
Table 5. ADME analysis of top ten MPP phytochemical inhibitors by using SwissADME.
DISCUSSION
SARS-CoV-2 currently represents a global challenge for the scientific community, as the pandemic impact dangerously affects millions of people and kills thousands of lives every day. However, to date, no satisfactory progress has been made in the treatment of SARS-CoV-2 [67-70]. Several attempts have been made to treat this disease, but these drug candidates are still questionable due to low efficacy [71].
Drug discovery rate is increasing with the aid of computational biology [72]. This sector is now broadly used in the biopharmaceutical industry to detect and develop new lead compounds against many infectious pathogens [72, 73]. Thus, it is possible to visualize the ability of potential small molecules to bind to ligands/inhibitors [74]. SARS-CoV-2 MPP shares 96% similarity with SARS- CoV MPP [18, 75]. CoV MPP is an essential viral protein for the viral life cycle and a potential target to prevent the spread of infection by inhibiting the viral polyprotein degradation [15]. The discovery of the COVID-19 MPP structure provides a fantastic opportunity to identify potential drug candidates for treatment. Paritaprevir [76], nelfinavir [77] had already been accepted for the treatment of HIV or HCV. Lopinavir and ritonavir are protease inhibitors previously recommended for the treatment of SARS and MERS, which have similar mechanisms of action as HIV [78]. In our study, paritaprevir and lopinavir were used as comparative drug standards. Several compounds, such as flavonoids, have been reported to show antiviral bioactivities [79-82]. We investigated, azadirachtin, -12.5kcal/mol; rutin, -9 kcal/mol; theaflavin, -9 kcal/mol; astragalin, -8.8 kcal/mol have a higher binding affinity than the control paritaprevir, -8.7 kcal/mol and lopinavir, -7.9 kcal/mol. Azadirachtin forms non-covalent bonds with the crucial catalytic residue His-41 and Cys-145. However, rutin forms one hydrogen bond with Cys-145 and a non-covalent bond with His-41, whereas theaflavin forms one hydrogen and alkyl bonds with Cys-145. Thereby, these may act as inhibitors of SARS-CoV-2 main protease. The selected four compounds form strong non-covalent interactions with other binding site residues. However, similar recent studies also support these findings where the inhibitor compounds form strong covalent and non-covalent bonds with the following residues His-41, Met-49, Tyr-54, Phe-140, His-164, Met-165, Glu-166, Pro-168, Asp-187, Arg-188, and Gln-189 [82, 83]. There have been previous reports of in vitro and in vivo inhibitory potential for crude aqueous Neem leaf extract and the pure neem compound (azadirachtin) in dengue type 2 virus replication [42]. Several studies have shown that rutin has important pharmacological activities, including anti-inflammation, anti-oxidation, anti-adipogenic, neuroprotective, anti-diabetic, and hormone therapy [84]. It also has antiviral and immunomodulatory effects on dengue virus [85]. Theaflavin has antiviral activity against herpes simplex viral infections [86], an inhibitor of HCV entry, and promising for the development of a therapeutic arsenal for HCV infection [87]. It has also verified anti-influenza virus and anti-inflammatory activities [88]. Astragalin has effective medicinal activities like antioxidant, anti-inflammatory, cardioprotective, anticancer, antiobesity, neuroprotective, antiosteoporotic, antidiabetic, and antiulcer properties [45]. The binding sites for each ligand occupy the catalytic domain of SARS-CoV-2 main protease protein [46]. Of the usual binding residues, His-41 and Cys-145 form the catalytic dyad and function as substrate recognition sites [45, 89]. The top candidates were well fitted into the active pocket of MPP where several hydrophobic amino acid residues including Met-49, Gly-143, Cys-145, Met-165, Pro-168, Ala-191 form a relatively hydrophobic environment that can help stabilize the conformation [89]. In silico ADMET analysis is a productive, comprehensive, fast and economical way to test the physicochemical and pharmacological properties of each compound [90]. This analysis provides a clear image of potential drug candidates. Therefore, the best drug candidates were employed for ADME analysis to examine drug profiles. However, none of the metabolites showed side effects that could reduce their medicinal properties. SARS-CoV-2 manifests itself as a severe acute respiratory disease rather than a neuro disease [91]. Therefore, it is not necessary to cross the blood brain barrier (BBB) to be effective against SARS-CoV-2. Thus, no BBB permeants were found among the best drug candidates. Drug interaction with cytochrome P450 (CYP) is crucial for drug discovery. It is now accepted that many drug interactions can be explained by changes in metabolic enzymes found in the liver and other extra-hepatic tissues. Standard drug doses may cause detrimental effects related to elevated drug serum levels if a person is a poor metabolizer or has a CYP450 enzyme inhibitor added to therapy [92]. The study of cytochromes P450 (CYP) isoforms inhibition concluded that the suggested MPP inhibitors had fewer possibilities to interact with cytochromes P450 (CYP) isoforms.
CONCLUSIONS
COVID-19 has created a catastrophic global crisis affecting thousands of people every day, having already claimed thousands of lives, and severely hampered the global economy. As a contribution to this fight against SARS-CoV-2, virtual screening based molecular docking was carried out to identify new compounds that could bind the MPP of COVID-19. Our study proposes that phytochemicals such as azadirachtin, rutin, theaflavin and astraglin have a better binding affinity to MPP of COVID-19 than paritaprevir and lopinavir. Further in vitro and in vivo analyses are required to transform these potential inhibitors into clinical drugs. We anticipate that the insights obtained in the present study may prove valuable for exploring and developing novel natural anti- SARS-CoV-2 therapeutic agents in the future.
ACKNOWLEDGEMENT
None.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
SF conceived the idea and collected all information. SF, NRS, MH and NAK participated in the idea development, analyzed data and prepared the original draft. MSH reviewed and edited the manuscript. All authors read and approved the final version of the manuscript.
References
- [1]Parvez M, Alam S, Karim M, Hasan M, Jaman J, Karim Z, Tahsin T, Hasan M, Hosen MJ. Prediction of potential inhibitors for RNA-dependent RNA polymerase of SARS-CoV-2 using comprehensive drug repurposing and molecular docking approach. Int J Biol Macromol. 2020: 1787–1797.
- [2]Worldometer, Coronavirus Cases, Worldometer, 2020: 1–22.
- [3]WHO, Coronavirus Disease (COVID-19) Situation Dashboard, 2020.
- [4]Porcheddu R, Serra C, Kelvin D, Kelvin N and Rubino S. Similarity in case fatality rates (CFR) of COVID-19/SARS-COV-2 in Italy and China. J Infect Dev Count. 2020: 125–128.
- [5]Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G, Ruan, L, Song B, Cai Y, Wei M et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl J Med. 2020; 382: 1787–1799.
- [6]Tripp RA, Tompkins SM, editors. Roles of Host Gene and Non-coding RNA Expression in Virus Infection. Springer; 2018 Dec 24.
- [7]Lu H. Drug treatment options for the 2019-new coronavirus (2019-nCoV). Biosci Trends 2020; 14(1): 69-7.
- [8]Li G, De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019- nCoV). Nat Rev Drug Discov. 2020; 19(3):149–50.
- [9]Pal M, Berhanu G, Desalegn C, Kandi V. Severe acute respiratory syndrome Coronavirus-2 (SARS-CoV-2): An update. Cureus. 2020; 12(3).
- [10]Kannan S, Shaik Syed Ali P, Sheeza A, Hemalatha K. COVID-19 (Novel Coronavirus 2019) – recent trends. Eur Rev Med Pharmacol Sci 2020; 24(4): 2006–11.
- [11]Mousavizadeh L, Ghasemi S. Genotype and phenotype of COVID-19: their roles in pathogenesis. J Microbiol Immunol Infect. 2020; S1684–1182(20):30082–7.
- [12]Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. 92; 92(4): 418–423.
- [13]Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and corona virus disease-2019 (COVID-19): the epidemic and the challenges. Int J Antimicrob Agents. 2020: 105924.
- [14]Xia B, Kang X. Activation and maturation of SARS-CoV main protease. Protein & cell. 2011; 2(4): 282-90.
- [15]Liu X, Zhang B, Jin Z, Yang H, Rao Z. The crytal structure of 2019-nCoV main protease in complex with an inhibitor N3. RCSB Protein Data Bank. 2020.
- [16]Zhou M, Zhang X, Qu J. Coronavirus disease 2019 (COVID-19): a clinical update. Front Med 2020; 2:1–10.
- [17]Lu H. Drug treatment options for the 2019-new coronavirus (2019-nCoV). Biosci Trends. 2020.
- [18]Xu Z, Peng C, Shi Y, Zhu Z, Mu K, Wang X, Zhu W. Nelfinavir was predicted to be a potential inhibitor of 2019-nCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. BioRxiv. 2020.
- [19]Koc Suheda, Belgin S, Isgor, Yasemin G, Isgor, Moghaddam NS, et al. The potential medicinal value of plants from Asteraceae family with antioxidant defense enzymes as biological targets. Pharmaceut Biol 2015; 53(5):746–51.
- [20]Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Duan Y, Yu J. Structure-based drug design, virtual screening and high-throughput screening rapidly identify antiviral leads targeting COVID-19. BioRxiv. 2020.
- [21]Lee TW, Cherney MM, Huitema C, Liu J, James KE, Powers JC, Eltis LD, James MN. Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate-like aza-peptide epoxide. J Mol Biol. 2005; 353(5): 1137-51.
- [22]Kankanamalage AC, Kim Y, Rathnayake AD, Damalanka VC, Weerawarna PM, Doyle ST, Alsoudi AF, Dissanayake DP, Lushington GH, Mehzabeen N, Battaile KP. Structure-based exploration and exploitation of the S4 subsite of norovirus 3CL protease in the design of potent and permeable inhibitors. Eur J Med Chem. 2017; 126: 502-16.
- [23]Berman HM, Bourne PE, Westbrook J and Zardecki C. The protein data bank. In Protein structure. CRC Press. 2003: 394–410.
- [24]Thompson JD, Gibson TJ, Higgins DG. Multiple Sequence Alignment Using ClustalW and 26 ClustalX. Curr Protoc Bioinforma. 2003; 00:2.3.1-2.3.22.
- [25]Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018; 35:1547–9.
- [26]Seeliger D, de Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol. 2010; 24(5): 417-22.
- [27]Liu X, Zhang B, Jin Z, Yang H, Rao Z. The crytal structure of 2019-nCoV main protease in complex with an inhibitor N3. RCSB Protein Data Bank. 2020.
- [28]Biovia DS. Discovery studio modeling environment. 2017.
- [29]Guex N, Peitsch MC. SWISS‐MODEL and the Swiss‐Pdb Viewer: an environment for comparative protein modeling. Electrophoresis. 1997; 18:2714-23.
- [30]Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004; 25:1605-12.
- [31]O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminformatics. 2011; 3:33.
- [32]Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Biol. 2015; 1263:243-50.
- [33]Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem. 1996; 17:490-519.
- [34]Li Y, Zhang J, Wang N, Li H, Shi Y, Guo G and Zou Q. Therapeutic Drugs Targeting 2019-nCoV Main Protease by High-Throughput Screening. bioRxiv. 2020
- [35]RA L, MB S. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011; 51: 2778–86.
- [36]Daina A, Michielin O, Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017; 7: 1–13.
- [37]Daina A, Zoete V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem. 2016: 1117–21.
- [38]K. Anand , J. Ziebuhr , P. Wadhwani , J. R. Mesters and R. Hilgenfeld , Coronavirus main proteinase (3CLpro) Structure: Basis for design of anti-SARS drugs. Science. 2003; 300: 1763 —1767
- [39]Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y, Jung SH. An overview of severe acute respiratory syndrome–coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016; 59: 6595 —6628.
- [40]Klibanov OM, Gale SE, Santevecchi B. Ombitasvir/paritaprevir/ritonavir and dasabuvir tablets for hepatitis C virus genotype 1 infection. Ann Pharmacother. 2015; 49(5): 566-81.
- [41]van der Leur MR, Burger DM, la Porte CJ, Koopmans PP. A retrospective TDM database analysis of interpatient variability in the pharmacokinetics of lopinavir in HIV-infected adults. Ther Drug Monit. 2006; 28(5): 650-53.
- [42]Rahmani A, Almatroudi A, Alrumaihi F, Khan A. Pharmacological and therapeutic potential of neem (Azadirachta indica). Pharmacogn Rev. 2018; 12(24).
- [43]Patel K, Patel DK. The beneficial role of rutin, a naturally occurring flavonoid in health promotion and disease prevention: a systematic review and update. In Bioactive Food as Dietary Interventions for Arthritis and Related Inflammatory Diseases. 2019: 457-479.
- [44]Sahoo M, Jena L, Rath SN, Kumar S. Identification of suitable natural inhibitor against influenza A (H1N1) neuraminidase protein by molecular docking. Genomics Inform. 2016; 14(3): 96.
- [45]Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, Sun L, Mo L, Ye S, Pang H, Gao GF. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proceedings of the National Academy of Sciences. 2003 Nov 11; 100(23):13190-5.
- [46]Mgbeahuruike EE, Yrjönen T, Vuorela H, Holm Y. Bioactive compounds from medicinal plants: Focus on Piper species. S Afr J Bot. 2017; 112: 54-69.
- [47]Park JY, Han X, Piao MJ, Oh MC, Fernando PM, Kang KA, Ryu YS, Jung U, Kim IG, Hyun JW. Hyperoside induces endogenous antioxidant system to alleviate oxidative stress. J Cancer Prev. 2016; 21(1): 41.
- [48]Tao Y, Zhan S, Wang Y, Zhou G, Liang H, Chen X, Shen H. Baicalin, the major component of traditional Chinese medicine Scutellaria baicalensis induces colon cancer cell apoptosis through inhibition of oncomiRNAs. Sci Rep. 2018; 8(1): 11.
- [49]Balandrin MF. Commercial utilization of plant-derived saponins: an overview of medicinal, pharmaceutical, and industrial applications. In Saponins used in traditional and modern medicine. Springer, Boston, MA. 1996: 1-14.
- [50]Esposito F, Carli I, Del Vecchio C, Xu L, Corona A, Grandi N, Piano D, Maccioni E, Distinto S, Parolin C, Tramontano E. Sennoside A, derived from the traditional chinese medicine plant Rheum L., is a new dual HIV-1 inhibitor effective on HIV-1 replication. Phytomedicine. 2016; 23(12): 1383-91.
- [51]Gokulan K, Kolluru P, Cerniglia CE, Khare S. Dose-dependent effects of Aloin on the intestinal bacterial community structure, short chain fatty acids metabolism and intestinal epithelial cell permeability. Front Microbiol. 2019; 10: 474.
- [52]Chu C, Deng J, Man Y, Qu Y. Green tea extracts epigallocatechin-3-gallate for different treatments. BioMed Research International. 2017.
- [53]Burdick EM. Carpaine: An alkaloid of Carica papaya: Its chemistry and pharmacology. Econ Bot. 1971: 363-5.
- [54]Houghton PJ, Woldemariam TZ, Watanabe Y, Yates M. Activity against Mycobacterium tuberculosis of alkaloid constituents of Angostura bark, Galipea officinalis. Planta Med. 1999; 65(03): 250-4.
- [55]He M, Min JW, Kong WL, He XH, Li JX, Peng BW. A review on the pharmacological effects of vitexin and isovitexin. Fitoterapia. 2016; 115: 74-85.
- [56]Fazal F, Mane PP, Rai MP, Thilakchand KR, Bhat HP, Kamble PS, Palatty PL, Baliga MS. The phytochemistry, traditional uses and pharmacology of Piper Betel. linn (Betel Leaf): A pan-asiatic medicinal plant. Chin J Integr Med. 2014:1-11.
- [57]Salehi B, Venditti A, Sharifi-Rad M, Kręgiel D, Sharifi-Rad J, Durazzo A, Lucarini M, Santini A, Souto EB, Novellino E, Antolak H. The therapeutic potential of apigenin. Int J Mol Sci. 2019; 20(6): 1305.
- [58]Mitrocotsa D, Mitaku S, Axarlis S, Harvala C, Malamas M. Evaluation of the antiviral activity of kaempferol and its glycosides against human cytomegalovirus. Planta Med. 2000; 66(04): 377-9.
- [59]Ding Y, Cao Z, Cao L, Ding G, Wang Z, Xiao W. Antiviral activity of chlorogenic acid against influenza A (H1N1/H3N2) virus and its inhibition of neuraminidase. Sci Rep. 2017; 7: 45723.
- [60]Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y, Jung SH. An overview of severe acute respiratory syndrome–coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J Med Chem. 2016; 59(14): 6595-628.
- [61]Pillaiyar T, Meenakshisundaram S, Manickam M, Sankaranarayanan M. A medicinal chemistry perspective of drug repositioning: Recent advances and challenges in drug discovery. Eur J Med Chem. 2020: 112275.
- [62]Tang B, Wang X, Li Q, Bragazzi NL, Tang S, Xiao Y, Wu J. Estimation of the transmission risk of the 2019-nCoV and its implication for public health interventions. J Clin Med. 2020; 9(2): 462.
- [63]Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C, Duan Y. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020:1-5.
- [64][64] Kumar V, Dhanjal JK, Kaul SC, Wadhwa R, Sundar D. Withanone and caffeic acid phenethyl ester are predicted to interact with main protease (Mpro) of SARS-CoV-2 and inhibit its activity. Journal of Biomolecular Structure and Dynamics. 2020:1-7.
- [65]da Silva Hage-Melim LI, Federico LB, de Oliveira NK, Francisco VC, Correa LC, de Lima HB, Gomes SQ, Barcelos MP, Francischini IA. Virtual screening, ADME/Tox predictions and the drug repurposing concept for future use of old drugs against the COVID-19. Life Sciences. 2020:117963.
- [66]Daina A, Zoete V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem. 2016: 1117–21.
- [67]Lake MA. What we know so far: COVID-19 current clinical knowledge and research. Clinical Medicine. 2020; 20(2): 124.
- [68]Yuen KS, Ye ZW, Fung SY, Chan CP, Jin DY. SARS-CoV-2 and COVID-19: The most important research questions. Cell Biosc. 2020; 10(1): 1-5.
- [69]Fang Y, Zhang H, Xie J, Lin M, Ying L, Pang P. Sensitivity of chest CT for COVID-19: comparison to RT-PCR. Radiology. 2020.
- [70]Dong L, Hu S, Gao J. Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discov Ther. 2020; 14: 58–60.
- [71]Dan Z, Sheng-Ming D, Qiang T. COVID-19: a recommendation to examine the effect of hydroxychloroquine in preventing infection and progression| Journal of Antimicrobial Chemotherapy. J Antimicrob Chemother. 2020:1-4.
- [72]Hirono S. An introduction to the computer-aided structure-based drug design–applications of bioinformatics to drug discovery. Rinsho byori. 2002; 50(1): 45-51.
- [73]Ivanov AS, Veselovsky AV, Dubanov AV, Skvortsov VS. Bioinformatics platform development: from gene to lead compound. Methods Mol Biol. 2006; 316: 389-431.
- [74]Joseph J, Bhaskaran R, Kaliraj M, Muthuswamy M, Suresh A. Molecular Docking of Phytoligands to the viral protein receptor P. monodon Rab7. Bioinformation. 2017; 13(4): 116.
- [75]Zhavoronkov A, Zagribelnyy B, Zhebrak A, Aladinskiy V, Terentiev V, Vanhaelen Q, Bezrukov DS, Polykovskiy D, Shayakhmetov R, Filimonov A, Bishop M. Potential non-covalent SARS-CoV-2 3C-like protease inhibitors designed using generative deep learning approaches and reviewed by human medicinal chemist in virtual reality. 2020.
- [76]Sulkowski MS, Eron JJ, Wyles D, Trinh R, Lalezari J, Wang C, Slim J, Bhatti L, Gathe J, Ruane PJ, Elion R. Ombitasvir, paritaprevir co-dosed with ritonavir, dasabuvir, and ribavirin for hepatitis C in patients co-infected with HIV-1: a randomized trial. Jama. 2015; 313(12): 1223-31.
- [77]Regazzi M, Maserati R, Villani P, Cusato M, Zucchi P, Briganti E, Roda R, Sacchelli L, Gatti F, Delle Foglie P, Nardini G. Clinical pharmacokinetics of nelfinavir and its metabolite M8 in human immunodeficiency virus (HIV)-positive and HIV-hepatitis C virus-coinfected subjects. Antimicrob Agents Chemother. 2005; 49(2): 643-9.
- [78]Li JY, You Z, Wang Q, Zhou ZJ, Qiu Y, Luo R, Ge XY. The epidemic of 2019-novel-coronavirus (2019-nCoV) pneumonia and insights for emerging infectious diseases in the future. Microb Infect. 2020; 22(2): 80-85.
- [79]Zakaryan H, Arabyan E, Oo A, Zandi K. Flavonoids: promising natural compounds against viral infections. Arch Virol. 2017; 162(9): 2539-51.
- [80]Thayil SM, Thyagarajan SP. Pa-9: A flavonoid extracted from plectranthus amboinicus inhibits HIV-1 protease. Int J Pharmacogn Phytochem Res. 2016; 8(6):1020-4.
- [81]Jo S, Kim S, Shin DH, Kim MS. Inhibition of SARS-CoV 3CL protease by flavonoids. J Enzyme Inhib Med Chem. 2020; 35(1): 145-51.
- [82]Dai W, Zhang B, Jiang XM, Su H, Li J, Zhao Y, Xie X, Jin Z, Peng J, Liu F, Li C. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020; 368(6497): 1331-35.
- [83]Jin Z, Zhao Y, Sun Y, Zhang B, Wang H, Wu Y, Zhu Y, Zhu C, Hu T, Du X, Duan Y. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat Struct Mol Biol. 2020; 27: 529-32.
- [84]Chua LS. A review on plant-based rutin extraction methods and its pharmacological activities. J Ethnopharmacol. 2013; 150(3): 805-17.
- [85]Keivan Z, Teoh BT, Sam SS, Wong PF, Mustafa MR, AbuBakar S. In vitro antiviral activity of fisetin, rutin and naringenin against dengue virus type-2. J Med Plants Res. 2011; 5(23): 5534-39.
- [86]De Oliveira A, Prince D, Lo CY, Lee LH, Chu TC. Antiviral activity of theaflavin digallate against herpes simplex virus type 1. Antivir Res. 2015; 118: 56-67.
- [87]Chowdhury P, Sahuc ME, Rouillé Y, Rivière C, Bonneau N, Vandeputte A, Brodin P, Goswami M, Bandyopadhyay T, Dubuisson J, Séron K. Theaflavins, polyphenols of black tea, inhibit entry of hepatitis C virus in cell culture. PloS One. 2018; 13(11): e0198226.
- [88]Zu M, Yang F, Zhou W, Liu A, Du G, Zheng L. In vitro anti-influenza virus and anti-inflammatory activities of theaflavin derivatives. Antiviral Res. 2012; 94(3): 217-24.
- [89]Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, Wang Q, Xu Y, Li M, Li X, Zheng M. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharmaceutica Sinica B. 2020.
- [90]Talele TT, Khedkar SA, Rigby AC. Successful applications of computer aided drug discovery: moving drugs from concept to the clinic. Curr Top Med Chem. 2010; 10: 127-41.
- [91]Astuti I. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes & Metabolic Syndrome: Clinical Research & Reviews. 2020.
- [92]Meyer UA. Pharmacogenetics and adverse drug reactions. The Lancet. 2000; 356(9242): 1667-71.