Impact of process validation and equipment qualification in production of bio-therapeutics
Abstract
Production of therapeutic proteins like Biosimilars uses a complex cell based fermentation and purification process. There are chances of errors in these complex production processes due to complexities in the molecular pathway that every protein follows. Therefore to produce a desired therapeutic protein that elicit the correct immunological response in the patient, one need to ensure that it must have correct sequence, size, structure so that they are recognized by the specific receptors. The quality of these therapeutic proteins is characterized through standard assays and methods such as SDS PAGE (sodium dodecyl sulphate–polyacrylamide gel electrophoresis), IEF (Isoelectric focusing), Western Blot and other methods those are based on HPLC, Mass spectrometry and capillary electrophoresis to conform the desired molecular size, purity and identity of proteins. Thus in the process of validation of a method, equipment plays a critical role in ensuring the quality and safety of a biopharmaceutical product. It is always necessary to generate scientific data and information such as IQ (Installation qualification), OQ (Operational qualification) and PQ (Performance qualification) protocol and reports for each instrument employed in the process at the beginning and before the actual testing of the desired product. It is only after the completion of these activities, the product under different manufacturing steps can be characterized. Involvement of a software in conjunction to an equipment is also common and it is important that the software is in use with the equipment also meets the requirements of the CFR 21 FDA (Code of Federal Regulations, 21 Part 11 of US Food and Drug Administration), which is a legal requirement in a modern manufacturing process and hence it is important to comply with these rules as set of guidelines that will help to ensure the integrity and safety of data.
Keywords
INTRODUCTION
The fundamental goal of any pharmaceutical industry is to reliably deliver the product of the necessary quality and at the most minimal conceivable expense. Validation has for quite some time been a significant process in the pharmaceutical industry, yet it has gotten more consideration lately as the industry turns out to be more worried about quality confirmation and improving efficiency [1].
The words ‘validation’ and ‘qualification’ are often mixed up and used interchangeably leading to misleading their actual sense of scope. The word “validation’ originates from the Latin word “validus” somewhere in the mid-17th century and is best described as the process of establishing material evidence by virtue of documentation. Validation is an archived demonstration of showing that a process, procedure, device, material, action or framework can really lead to an expected results [2, 3]. Validation gives adaptability in controlling the manufacturing process to accomplish the desired properties in a therapeutic protein / drugs while keeping away from undesirable one [4]. This essentially assures that a specific process will show consistency to produce a product which will meet its predetermined specifications and quality [5]. The word qualification also originates from Medieval Latin of “qualificare” and is described as the act or process to assure that something complies with an expected outcome, standard or a set of specific requirements. There are three distinct phases in a validation process [6, 7].
The purpose of this paper is to succinctly review the recent progresses in validation process and the understanding on how to minimize the risk of failure of a product in manufacturing in biopharmaceutical industry. Thus the current article addresses the steps involves in this validation process of a technology and the role of equipment qualification in ensuring that it will deliver the desired quality of the product.
PRE-VALIDATION PHASE OR QUALIFICATION PHASE
It covers all activities relating to product in research and development, formulation of pilot batch studies, scale-up studies, transfer of technology to commercial scale batches, establishing stability conditions, storage and handling of in-process and finished dosage forms, equipment qualification, installation qualification, master production document, operational qualification and process capacity [8].
PROCESS VALIDATION PHASE OR PROCESS QUALIFICATION PHASE
It is designed to verify that all established limits of the critical process parameters are valid and that satisfactory products can be produced even under the “worst case” conditions [9, 10].
VALIDATION MAINTENANCE PHASE
This phase requires frequent review of all process related documents, including validation of audit report to assure that there has been no changes, deviations, failures, modifications to the production process and that all SOPs have been followed, including change control procedure. At this stage the validation team also ensures that there is no change or deviations that should have resulted in requalification and revalidation [11].
Therapeutic proteins or as commonly referred as “biosimilar” are such biological products which are most routinely produced through cell culture process involving bioreactors. The amplified proteins after harvesting goes through a complex downstream process to get the desired purified product [12]. The mechanism through which these therapeutic proteins are obtained by harnessing the machinery of the cells is a very complex molecular pathways such as transcription and translation. The complexity of these processes are often prone to errors which can takes place at different levels of their production process. To deliver their desired functionality, it is mandatory after post translation modification for these therapeutic proteins to have a correct structure, size and sequence [13]. This is needed for their recognition by specific receptor(s) and should be able to elicit the correct immunological response in the patient. Thus, it is very important to monitor and ensure a correct and consistent structure, sequence, purity and stability during the production of these proteins. All aspects of the product thus obtained should undergo analysis at different stages during the processing steps right from the stages of discovery to the upstream and downstream process development leading to final therapeutic product [14].
Industries employ different techniques with varying complexities as characterization tools which involve simple methods like protein estimation assays to quantify the yield, Isoelectric Focusing (IEF) to determine the isoforms, Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis (SDS–PAGE) to analyse the size of the protein and Western Blot to check their identity and specificity. The other commonly used method are HPLC, Mass Spectrometry and Capillary Electrophoresis based [15, 16]. All these analysis provide vital data with respect to the identity, charge, molecular size and purity of these therapeutic proteins [17, 18]. Thus the process of validation of method and the instruments play a critical role in the quality and safety of the product [50]. It is necessary and important to have the qualification and validation of the instruments in order to obtain an authentic, reliable and repeatable analysis. Accordingly, before the analysis of these products by various methodologies and its validation, all laboratory instruments involved in the process must have performed four major qualification elements [19, 20], such as design qualification (DQ), installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ) [21,22]. Once the instruments are qualified, the technique or analytical methodologies must be validated according to ICH Q2 guidelines [23].
The concept of qualification is incorporated in validation (Figure 1) wherein the scope of validation study is larger and involves various qualifications to attend the end result. Thus, in validation or qualification, the requirement for establishing documented evidence in terms of the nature of activities supersedes its importance than the mere name of these activities [24, 25]. It is always necessary that at the beginning and prior to the actual testing of the desired product, the generation of scientific data and information, the IQ, OQ and PQ protocol and reports for the respective instruments are completed. A method validation protocol and report should also be generated for individual test methods prior to analysis of the test product. It is after completion of these activities, the product under different manufacturing steps can be characterized. Tests scan then be initiated to characterize the product through the manufacturing process. Involvement of a software in conjunction to an equipment is also common and it is important that this software also fulfil the provisions of CFR 21 FDA (Code of Federal Regulations, 21Part 11), which is a legal requirement and help ensure the reliability and protection of the data [25].
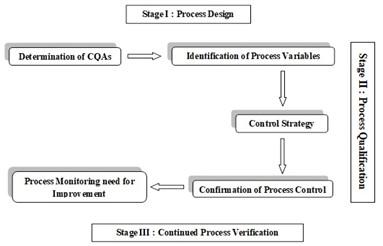
SCOPE AND IMPORTANCE OF VALIDATION
Modern day biotechnology provides discoveries of recombinant DNA technology to discover and develop therapeutic biologics such as monoclonal antibiotics, therapeutic vaccines, growth hormones, blood production-stimulating proteins, therapeutic enzymes, cytokines, recombinant proteins, Insulin and its analogs, gene therapy, fusion proteins, biosimilars to name a few [26, 27].
Manufacturing scale bioreactors are used to produce intended therapeutic proteins from specific genetically engineered cells. This is then harvested and further purified for final product. The culture cycle and protein production in the cell occurs through a complex mechanism involving DNA transcription in the nucleus and the protein translation in the cytoplasm. The final biologically active protein is obtained through folding and post-translational modifications of the initial polypeptide chains. During these complex cellular processes, a variety of errors are encountered at several stages during the cellular process. A correct size, a correct structure, and a correct sequence followed by an accurate post-translational modifications is mandatory for a desired effect to be recognized by their intended binding partner. A biologically active therapeutic protein with proper physiochemical and immunochemical properties can only yield the appropriate beneficial response for the ailing person’s intended therapy and to achieve accuracy in such critical product development, the process has to run through a robust validation process involving following steps [28, 29, 30].
- Quality assurance.
- Process optimization.
- Validate commercially reproducible process designs and acquire knowledge into the process.
- To spot the worst cases and risks that may arise during the production of quality products.
- Validation to examine the deviations caused during the process.
- Variability within and between batches can be evaluated.
- Reduce the production cost of the products.
- Avoidance of capital expenditures.
All the above attributes are commonly complied within the various regulatory requirement such as GMP (good manufacturing practices), FDA (Food and Drug Administration, USA), MHRA (medicines and healthcare products regulatory agency, UK), TGA (therapeutic goods administration, Australia) and other regulatory agencies) [31, 32]. Therefore, monitoring, controlling and ensuring the purity of a therapeutic protein, its structural sequence, size and stability play a vital role during production of such bio-molecules [51]. Any errors with the above mentioned parameters with respect to the therapeutic protein could fail in eliciting beneficial effects to the patient, or in worse case, these inaccuracies could affect the safety of the drug and have detrimental affect resulting in an undesired immunogenic response which may lead to very dangerous anaphylaxis shock and even death [24, 33, 34].
STEPS INVOLVED IN BIOLOGICAL PRODUCT DEVELOPMENT
A typical biologic product development milestone involves proof of concept followed by process development which is further subdivided into upstream, downstream and analytical product characterization followed by engineering run which includes material generation for toxicology, animal studies (safety), reference standard preparations (efficacy) and stability studies (process validation) which are clubbed under pre-clinical stages[28, 29]. The pre-clinical stage is followed by the clinical studies which include production of clinical grade material under GMP conditions to support phase 1, phase 2 and phase 3 trials [25, 35, 36]. The data arising from pre-clinical and clinical studies are submitted separately to the regulatory bodies for review and further approvals for manufacturing at a commercial scale [30].
THE REGULATORY AUTHORITIES
There are several guidance documents provided by regulatory bodies such as FDA, MHRA, TGA and others help industries to analyze the product during intermediate stages of the development which include the discovery stage, the upstream processing stages, downstream or purification stages till the final polished product is obtained [31, 37, 38]. These analyses the product at various stages of its manufacture, once incorporated into the production system will ensures that the product is reliable, stable, safe and pure from start to finish [39, 40].
Regulatory agencies in leading countries are named differently and are also vary in their continuously evolving stringency level. In United States it is United States food and drug administration -USFDA, in Europe it is European medical agency –EMA [41, 42, 43], in Canada they have – Health Canada, in Australia it is the therapeutic goods administration – TGA, in China, it is China’s state food and drug administration. In India the primary regulatory agency for drugs and pharmaceutical is known as central drugs standard control organization (CDSCO) under the Indian ministry of health and family welfare [44, 45]. Various nations that control biologics has their own regulations. However often there are opinions those are overlapping which may potentially create confusions. The international conference on harmonizing (ICH) Technical Requirements for the registration of pharmaceuticals for human use was formed in 1990 to unite these various regulatory bodies. The role of this organization is to bring together these regulatory authorities to discuss scientific and technical aspects of drug registration and harmonized regulatory guidance documents to help biopharmaceutical companies to register and develop safe, quality and effective drugs. These documents involve guidelines encompassing quality, safety and efficacy [33, 53].
ASPECTS
To ensure production meets quality standards, it is mandatory to have calibrated equipment, monitoring and testing of in-process functionalities, personnel training, development of SOPs, and maintenance of log book, batch production and control record. These are documented as quality standards that enhance the potential for monitoring a validated process. Equipment used for any process or analysis of the product should be installed according to its requirements. The calibration, maintenance and cleaning protocol developed for respective instrument used in the process as well as testing has to be documented as SOPs followed by test conducted to ensure the instrument is operating correctly. This also includes an operator training manual.
INSTRUMENT QUALIFICATION
Four different phases are involved in qualification of an instrument (figure 2). Instead of a single and continuous process, the results are obtained from many discrete activities and these four activities including DQ – design qualification, IQ – installation qualification, OQ – operational qualification, and PQ – performance qualification.
Analytical Instrument Qualification (AIQ) is accepted widely within the community of users, manufacturers and quality assurance across industries and manufacturers. Manufacturing process validation also has these qualification phases originating at different stages of its orientations [46, 36, 47]. Thus, it is very crucial that the required AIQ activities are in place. Some AIQ activities may be carried out in one or the other qualification process and it is not mandatory for these individual activities to be captured under single qualification head, can be performed and reported under different subgroups under which the individual activity is performed or reported [48, 49, 52, 54].
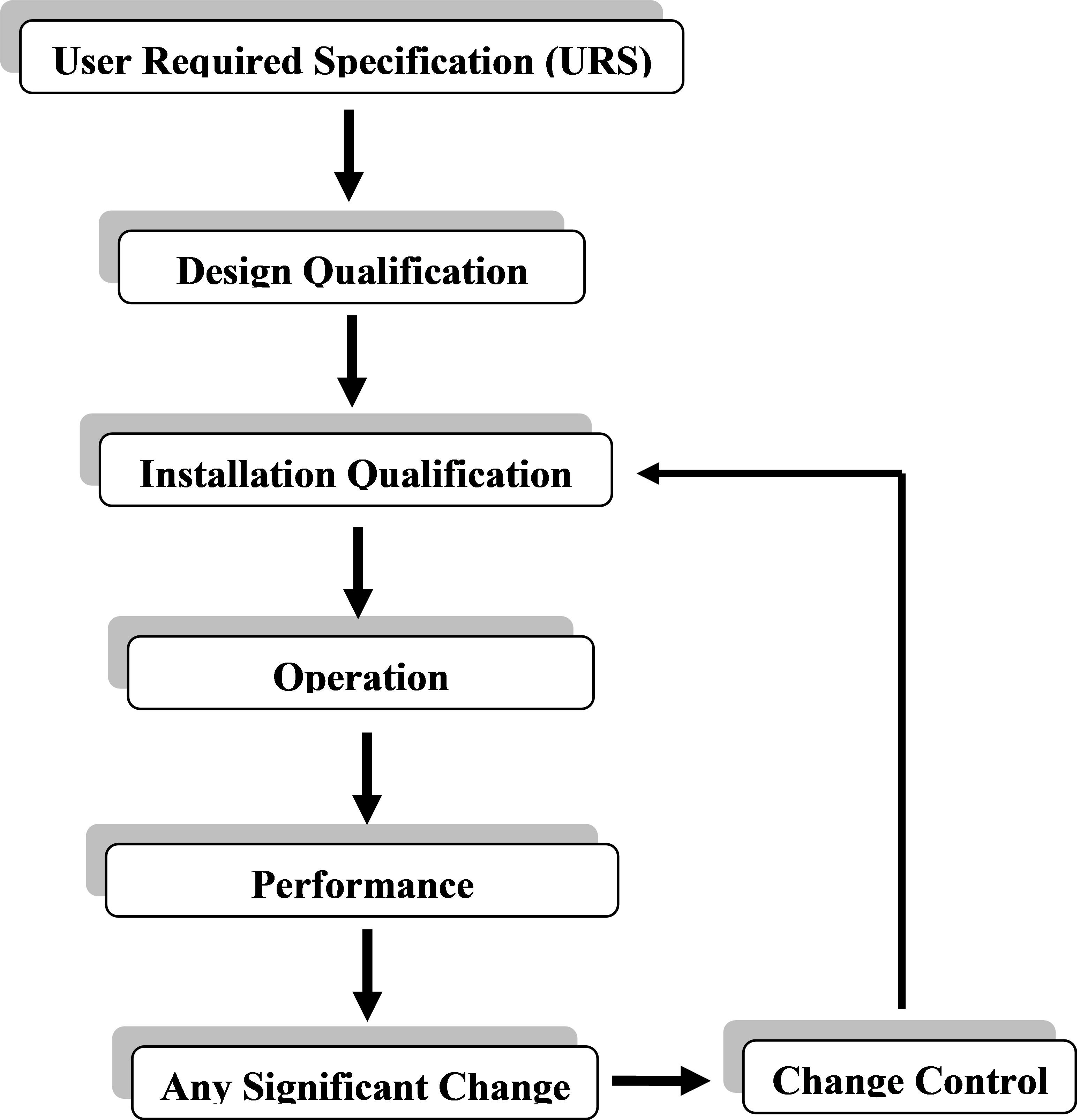
DQ- design qualification
The activity of design qualification is usually done by the developer or the manufacturer. Since the instrument design is already fixed for the commercial purchase, the user does not need to repeat all aspects of DQ. However, the general practice is that the users ensure that the manufacturer has adopted a system of quality for the development, production and testing of the instrument. It is to be checked that the manufacturers and vendors should adequately support installation, training, and services. The suitability of an instrument and its intended use depends on the functionality and design of the instrument and the complexity of the intended application for the instrument to perform. This is also extended to some extent to the user’s earlier experience with the manufacturer. The documents provided by the vendor also play a critical role for DQ purpose. The support quality and services given by vendors and the information of various instruments with respect to the suitability and its design attributes are often gathered through peer level technical interactions. A common practice also includes an informal visit to other users and/or vendor’s premises to analyze representative samples with specific instrument to determine the suitability of the instrument with respect to the intended use. The selection of an instrument becomes much easier a task if a due diligence of vendor support can be gathered through discussions with peer users.
IQ-installation qualification
A collection of documented activities performed during installation of an instrument at the user’s premises in termed as Installation Qualification. This is performed on a new or pre-owned instrument. It can also be performed on an existing instrument present onsite but had no previous records of IQ. IQ involves the following associated activates and its documentations:
- Description of the instrument: It is a set of documents which describes the instrument in terms of its design, the model number, serial number and the software details with the current version it runs on etc. and in some cases drawings or flowcharts are used wherever necessary.
- Delivery of the instrument: This is to ensure that the purchase order components like the instrument, its user manual, software and other accessories as specified in the PO should reach at the user’s end undamaged. Documents such as user manuals should be obtained for pre-owned instruments.
- Installation location: A place identified and selected for the instrument to be installed should satisfactorily meet the vendor requirement as per the working environment of the instrument. This requires a common judgment and understanding of the standard voltage that runs through the power ports in the installation area. Temperature and humidity condition of the area is also critical and should be in accordance with the working condition of the instrument to be installed.
- Network connection and storage of data: There are instruments which require network connection for data storage and further accessibility of these data to the user. If the instrument requires a network connection, then it is a must feature required at the installation site. The instrument should always be connected to the network and checked for its functionality.
- Assembly and installation: An initial diagnostics and further testing can be performed by the user after its assembly if a less complex instrument is involved. Whereas, it is best to leave the assembling and installation of complex instruments to the qualified engineers specialized for this purpose as provided by the vendor. The acceptance of a complex instrument is guided by various installation tests performed by the vendor which in turn forms a standard reference and a documentation is necessary if any abnormality is observed during the process of assembly and installation. This process is also followed for instruments which are pre-owned or have not yet gone through this qualification.
- Installation verification: After installation of an instrument, preliminary diagnostics check and testing is performed. The installation is only considered successful when the test results are found to be within the acceptable specifications by the user or the installation engineer. It is after which the next phase of qualification are to be taken up.
OQ-operational qualification
If the instrument meets the criteria of a successful installation qualification, next it is checked for its OQ which consists of the following tests:
- Fixed parameters: This parameter involves the non-variable aspects of the instrument like its dimensions (height, length, width) and weight etc, which does not change through the life of the instrument. Once the values of these parameters are obtained, they are never repeated. If the user is satisfied that the vendor has supplied the instrument according to its specification, then he or she may waive this determination. However, the parameters can be checked at the user’s site if they insist on a confirmation of these parameters.
- Backup and archive: A written procedure should be in place for the test of storage, backup, and archiving with a primary aim of handling data security.
- Instrument functionality: User of the instrument should verify that it is operating at par with his requirement and as claimed by the manufacturer by selecting essential instrument parameters for testing at the beginning and periodically according to the instrument’s projected use. A trained individual should identify these specifications and perform suitability tests to ascertain if the instrument performs as per user requirements. There can be two modes for OQ tests, like complete and modular. In a modular setup, the testing for individual critical component of the instrument is tested. This type of testing helps in interchangeability of components waving off the requalification steps. However, complete tests, wherein the total system is accounted for, are acceptable as against modular testing. The instrument is now qualified for use in regulated samples testing post the successful operational qualification tests yields satisfactory results.
PQ-performance qualification
A performance qualification is proved by the instrument’s continued suitability for its desired use after the IQ and OQ has been completed. The following parameters are included in PQ:
-Performance checks: A single or a series of tests can be performed to confirm the satisfactory working of the instrument for its desired use. PQ are usually checked based on the user’s set tests for which the instrument was purchased for in the first place. Some of these tests may be same as accomplished during OQ, but the specifications for their outcome can be set in a different way.
It is not just that performance qualification is required for newly installed instruments but also is routinely performed on a working instrument. Hence, PQ specifications can be slightly flexible than OQ specifications. Performance qualification should exhibit the instrument’s operation for its intended applications and a trouble free experience for its user. Separate PQ tests should be performed on the instrument segregating them from the routine analytical testing. The tests performed for this can also be in two different setups of either modular or complete like the one done for OQ. However, an effective evaluation of the entire or complete instrument is preferred over the separate testing of individual modules. The ruggedness and criticality of the test to be performed define the testing frequency of this parameter. It can either be unscheduled, i.e, before each time the instrument is used or it may be scheduled, where the testing frequency is defined at intervals of weeks, months or year. The testing frequency also depends on the experience of the user with the instrument. Generally, an identified critical selected set of PQ tests are conducted each time so that a history of the instrument’s performance can be monitored, captured and documented. The performance suitability of the instrument is also integrated with routine testing of samples by setting up quality control checks and system suitability tests.
-Preventive maintenance: If the results of a PQ test do not meet the set specifications, then the instrument may require maintenance or repair. Thus, it is always recommended that a periodical preventive maintenance is in place [49].
-Standard operating procedure for operation, calibration, and maintenance: To monitor and maintain the calibration details of an instrument, a system of standard operating procedures should be in place which may include the use of a logbook, binder or electronic record as documental proof which is necessarily a good practice.
Therefore the IQ, OQ, and PQ protocols and their reports need to be prepared and accepted for each piece of equipment at the very beginning of the test methodologies to evaluate the product. After these activities are completed, the characterization of product can be initiated through different testing techniques for the samples from different stages of its manufacturing. The role of a software associated with an instrument is also significant and during the validation process it should be mandatory that it meets the requirements of the CFR 21 FDA (Code of federal regulations, 21 Part 11), which is a legal requirement and is also important to look at the rule as set of guidelines that will help ensure the integrity and safety of data [24].
The FDA (Food and drug administration) in 1997, released the 21 CFR Part 11 final rule whereby it was recommended that electronic technology should be utilized at every possible opportunity. There are two sections in this parameter such as Electronic Records and Electronic Signatures.
The traditional use of paper records are further extended to these digitized forms of documentation. The security of data along with hand written signatures are provided in paper records which in turn indicates that certain data is correct and verified and any deliberate illegal changes in the data or signatures are easily noticeable. A high level of confidence is reflected in a securely generated electronic record which is equivalent to that of paper records. An equivalent alternative is an electronic signature through which both the operator and the supervisor can authenticate a document like they would have done in the case for their paper counterparts. The uses of biometric authentication such as fingerprint identification, face recognition, iris scan, voice analysis or retinal scan devices are also permitted in this rule. A significant amount of advantage for data management and data recovery is obtained by means of electronic documentation forms. In order to have confidence in an electronic format of data, the FDA established the 21 CFR Part 11 rules that describe the necessary steps for their security [24, 43, 50].
Easiness of validation
Electronically integrated data significantly reduces the validation period and analysis by using controlled, integrated features to comply with the FDA’s 21 CFR Part 11. It provides data collection at all stages, locally and across the plant. The data on no occasion is lost as it can have multiple recorded copies and secure backup without increasing any cost as in case of paper documents which requires a steady supply of stationaries and space. A centralized stable system ensures that the client accounts and passwords are overseen from multiple sites.
Electronic Records
Electronic records are a safe mechanism that involves audit trails and values (incidents, operator behavior, login / log-out, operator notes, alarms and electronic signatures). This too offers information security by twofold, compacted, check-summed records and ensures intervals are scaled to a defined clock source through automatic time synchronization. Precise time stamps are the provision for the electronic copying of data for the archive while export facility providing human readable viewing of protected documents.
Electronic signatures
It offers user-specific authority-level access and all user activities can be configured to allow signing or allow both signing and authorization. The signature aspect controls and maintains unique user signature, minimum password length, automatic log-out, password expiry, automatic disabling and notification of failed login attempts, and ensures users does not remove their accounts and do not delete any previously uploaded documents.
Audit trail
Security managers can achieve a significant amount of cost savings and convenience of use which enables user accounts and passwords to be managed from different sites. An updated password change by the user on a local PC/ system can take effect automatically through all the systems that they have access at different locations.
The advantages include a common safety method across several ranges of items. Constant help with an in-built audit trail for 21 CFR Part 11 validations for different security zones provides automated version control and support for electronic signatures which can be modified by the user and delivered to a multiple systems in one and more locations [44].
CONCLUSION
In addition to the regulatory requirement, the validation of analytical procedures is a critical aspect for their efficient and reliable long-term application. For executing the performance of an analytical procedure correctly it is important that the analyst is able to identify the relevant parameters adequately so as to design the experimental validation studies appropriately and to define acceptance criteria.
Thus the process of validation of method and the instruments play a critical role in the quality and safety of the product. The certification and validation process is indeed important and demanding. Therefore, before validating the specific test methodologies, all laboratory equipment must be installed and certified (IQd), operationally certified (OQd), and certified based upon performance (PQd). The validation of analytical procedures is another integral part for an industry with respect to their product efficacy and safety and is covered in ICH Q2 guidelines.
Establishing an acceptable analytical method for the given product is of central importance as the acceptance criteria can be dependent on the test results. The reliability of the analytical test is thus most sought after. The consistency and reliability of a validated process is therefore to produce a quality product is very important for industries.
ACKNOWLEDGEMENTS
Authors would like to acknowledge the support provided by Panacea Biotec Ltd. a leading Biopharmaceutical company in India and GIET University for facilitating in the development of this review article.
AUTHOR CONTRIBUTIONS
GG for Conceptualization this review report based on his long & real-time experience in biopharmaceutical product development, GG and BD has shared their experience in process development and analytics. GG, BD and RPD has contributed in writing and original draft preparation, GG, BD and RPB has contributed in further review and editing. All authors have read and agreed to the published version of the manuscript.
CONFLICTS OF INTEREST
There is no conflict of interest among the authors.
References
- [1]Nash R A, Wachter AH. Pharmaceutical Process Validation, (3rd edn). Marcel Dekker, Inc, New York, USA. 2003.
- [2]Abdrhman M G. Validation as applied for Pharmaceutical Processes: Adv. Pharm. Edu. and Res.2015; 5(2): 77-86.
- [3]Sharma V, Rana AC, Seth N. Industrial process validation of solid dosages form-A review. Int Res J Pharm. 2013; 4(5): 67-70.
- [4]Varshney P, Shah M, Patel P, Rohit M. Different Aspects Involved In Process Validation. Innovare Journal of Science. 2013; 1(2): 16-19.
- [5]Wazade MB, Walde S R, Ittadwar AM. Process validation: an essential process in pharmaceutical industry. International Journal of Pharmaceutical Sciences and Research.2012; 3(9): 3007-3022.
- [6]Aswal N, Joshi P, Choudhary A, Kothiyal K. Prospective Validation of Paracetamol Tablet Dosage Form. International journal of pharmaceutical and chemical sciences. 2013; 2(3): 1416-1425.
- [7]A, Saini S. Process validation of solid dosage form: a review. International Journal of Research in Pharmacy and Science.2013; 3(2): 12-30.
- [8]Sharma V, Rana AC, Seth N. Industrial process validation of solid dosages form- A review. International Res J Pharm.2013; 4(5): 67-70.
- [9]Prasad K. Science and Risk Based Approach to the process Validation – link from quality by design to process validation. International Journal of Pharmaceutical Science and Research. 2016; 7(3): 914-929.
- [10]Chapman KG. Proposed Validation Standard VS-1. Journal of Validation Technology.2000; 6 (2):502–521.
- [11]Jain NK. A text book of Pharmaceutical Product development, CBS publisher.2005; 525-533.
- [12]Richard K, Gabriela K, Krista HC. Key Regulatory Guidelines for the Development of Biologics in the United States and Europe. Biological Drug Products: Development and Strategies. Wiley, First Edition.2014; 4:75-109.
- [13]Jenkins N. Modifications of therapeutic proteins: challenges and prospects. Cytotechnology.2007; 53 (1-3): 121-5.
- [14]Gronemeyer P, Ditz R, Strube J. Trends in Upstream and Downstream Process Development for Antibody Manufacturing. Bioengineering. 2014; 1 (4): 188-212.
- [15]Umut B, Sümeyra T, Yildirim E, Algin Y. An overview of analytical method validation. Universal Journal of Pharmaceutical Research.2020; 5 (1): 47-52.
- [16]Mahar P V. A pharmaceutical process validation: an Overview. International Journal of Pharmaceutical Research and Bio-Science. 2014; 3(4): 243-262.
- [17]Marceline M F, Steffen P,Hartmut S. Tools for the analysis and characterization of therapeutic protein species. Biosimilars. 2016; (6): 17-24.
- [18]Struble E B, Kirschbaum N, Liu J, Marszal E, Shapiro M. Characterization of Therapeutic Proteins. Protein Therapeutics.2016; 69-121.
- [19]Zameeruddin M, Kale S S, Jadhav S B, Kadam, V S, Chaware S S. Process validation of oral solid dosage form: tablet – an overview. World Journal of Pharmacy and Pharmaceutical Sciences.2015; 4(12):358- 373.
- [20]Kashid VA, Nikam VK, Somwanshi S B, Dolas R T, Dhamak K B, Gaware V M, Kotade K B. Brief overview of pharmaceutical facility validation. Journal of Current Pharma Research. 2014; 4 (2):1134-1137.
- [21]Gowrisankar D, Abbulu K, Bala SO, Sujana K. Validation and Calibration of Analytical Instruments. J Biomed Sci. and Res. 2010; 2(2): 89-99.
- [22]USP<1058> AIQ Risk-Based Instrument Qualification Guidelines. Lab compliance solutions. 2011.
- [23]Validation of Analytical Procedures: Text and Methodology; Q2 (R1); ICH Harmonised Tripartite Guideline.2005; 4: 1-13.
- [24]Cho KH, Kim JS, Jeon MS, Lee K, Chung MK, Chang WS. Basic Principles of the Validation for Good Laboratory Practice Institutes. Toxicol, Res.2009; 25 (1):1-8.
- [25]Michael H. Director of Product Management at Complion Inc, Presentation on Facilitating Compliances with 21 CFR Part 11. 2018; 1-13.
- [26]Shrivastava A, Tripathi N K. Recent Developments in Bioprocessing of Recombinant Proteins: Expression Hosts and Process Development. Frontiers in bioengineering and biotechnology.2019; 7: 420.
- [27]Patil R, Walther J. Continuous Manufacturing of Recombinant Therapeutic Proteins: Upstream and Downstream Technologies. Advances in biochemical engineering/biotechnology. 2018; 165: 277-322.
- [28]Validation of Solid Dosage Forms the FDA view.1989; 15(6-7): 1119-1133.
- [29]Guidelines for Process Validation of Pharmaceutical Dosage Forms. Saudi Food and Drug Authority, Kingdom of Saudi Arabia.2010.
- [30]Nandhakumar L, Dharmamoorthy G, Rameshkumar S, Chandrasekaran S. An overview of pharmaceutical validation: quality Assurance view point. International journal of research in pharmacy and chemistry. 2011; 1 (4): 1003-1014.
- [31]US Department of human and health services, Food and Drug Administration, Center for drug evaluation and research (CDER), Center for biologics evaluation and research (CBER), Center for veterinary medicine (CVM), Guidance for industry, Process Validation: General principles and practices.2008.
- [32]Gupta GD, Garg R, and Aggarwal S. Guidelines on general principles of Validation: solid, liquid and sterile dosage forms. 2008; 6(1): 28-33.
- [33]Varaprasad R K. Biotech regulation in India: Problems and promises. Biotechnol. J. 2009; 4: 306–309.
- [34]Kiranbala J, Princy A, Meenakshi B. A Review on Pharmaceutical Validation and Its Implications. International Journal of Pharmacy and Biological Sciences. 2018; 8 (2): 117-126.
- [35]FDA’s guidance for industry, Quality Systems Approach to Pharmaceutical Current Good Manufacturing Practice Regulations Food and Drug Administration.2006; 1061, 20852.
- [36]Basic Principles of GMP; Qualification and Validation; WHO. 2006; 4: 2-28.
- [37]Sruthi K, Raviteja MN, Vishal N G. Comparison of Global Regulatory Approvals for Biosimilar Products. Intl. Journal of PharmTech Research. 2013; 5 (3): 924-935.
- [38]FDA Guidance for industry, “Process validation: general principles and practice”. http://www.ema.europa.eu/ema/pages/includes/document/open_document.jsp,web Content Id=WC500002916.2017.
- [39]Guidance for Industry Process Validation: General Principles and Practices. Current Good Manufacturing Practices (CGMP), U.S. FDA.2011; 1: 1-22.
- [40]Guidance for Industry: Process Validation: General Principles and Practices. U.S. Department of Health and Human Services, Food and Drug Administration, Centre for Drug Evaluation and Research (CDER), Centre for Biologics Evaluation and Research (CBER), Centre for Veterinary Medicine (CVM). 2011.
- [41]John et al. The Biomanufacturing of Biotechnology Products; Biotechnology Product Development.2014; 26:351-385.
- [42]Devesh K, Ruchi B V, Diwaker D. An Overview of Analytical Instrument Qualification with Reference to Pharmaceutical Industry. Journal of Drug Delivery and Therapeutics. 2018; 8(5): 99-103.
- [43]Maher P, Verma A. Pharmaceutical Process Validation An overview. International Journal of Pharmaceutical research and Bio-Science.2014; 3(4): 243-262.
- [44]Guidance for Industry; Part 11, Electronic Records; Electronic Signatures. Scope and Application; U.S. Department of Health and Human Services, Food and Drug Administration. 2003.
- [45]u, C. F. Regulations on Biotechnological Research and Biologically Modified Products. Harvard Library, Office for Scholarly Communication. 2002; 1-36.
- [46]Ludwig H. Analytical instrument qualification and system validation. http://www.chem. Agilent.com/Library/primers/Public/5990-3288EN.pdf.2017.
- [47]Varshney P, Shah M, Patel P, Rohit M. Different Aspects Involved In Process Validation. Innovare Journal of Science.2013; 1(2): 16-19.
- [48]Surendra K B et al. Qualification of Analytical Instruments for Use in the Pharmaceutical Industry. A Scientific Approach, AAPS Pharm Sci Tech. 2004; 5 (1):1-8.
- [49]McElroy J. An Introduction to Analytical Instrument Qualification and Validation – Meeting FDA Expectations. Pharmaceutical Online. 2018.
- [50]Note for Guidance on Process Validation – The Europe Agency for Evaluation of Medicinal Products; CPMP/QWP/848/96; EMEA/CVMP/598/1999.
- [51]Yajun J W, Brian H. Analytical Characterization of Proteins and Peptides in Biological Drug Products: Development and Strategies.2013; 2.10: 285-323.
- [52]Satinder K. Pharmaceutical Process Validation: A CGMP Concept. (2012); 1269.
- [53]K Ho. Manufacturing Process of Biologics. Int. Conf. on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Afssaps, France. 2011.
- [54]Nash R A, Wachter A H. Pharmaceutical Process Validation. An International Third Edition, Revised and Expended, Marcel Dekkar, Inc., New York.2003; 17-40.