Syzygium aromaticum as a possible source of SARS-CoV-2 main protease inhibitors: Evidence from a computational investigation
Abstract
SARS-CoV-2, a new and fast circulating coronavirus strain, infected over 214 countries and territories worldwide and caused global health emergencies. The absence of appropriate medicines and vaccinations has further complicated the condition. SARS-CoV-2 main protease (Mpro) is crucial for its propagation, and it is considered a striking target. This study used several computational approaches to determine the probable antagonist of SARS-CoV-2 Mpro from bioactive phytochemicals of Syzygium aromaticum. A total of 20 compounds were screened through in silico approach. The molecular dynamics simulation studies were then carried out for further insights. We found crategolic acid, oleanolic acid, and kaempferol have considerable binding affinity and important molecular contacts with catalytic pocket residues, His41-Cys145. The pharmacological properties through ADMET analysis also showed that these compounds could be used as safe drug candidates. The molecular dynamics simulation study further confirmed these compound’s stability with Mpro. However, further detailed in-vitro and in-vivo analyses are compulsory to evaluate the real potentiality of identified compounds.
INTRODUCTION
A novel coronavirus strain was stated in late 2019 in Wuhan, China, called SARS-CoV-2, linked to lethal respiratory sickness in the patients [1]. The SARS-CoV-2 infection is termed as coronavirus disease 2019, which has created severe health issues and separated countries from one another. This disease triggered a global medical emergency and severely affecting international travel, tourism, and trade [2]. SARS-CoV-2 lies in the beta Coronavirus family [3], which has a similar sequence identity with its descendants SARS-CoV [4, 5].
The beta-coronaviruses synthesize ~800 kDa polyproteins, which are enzymatically sliced to synthesize several proteins. The papain-like protease (PLpro) and main protease (Mpro) causes the proteolysis of coronavirus proteins [6]. The Mpro slices the polyprotein and generates various polypeptides vital for viral replication, transcription, and translation [4, 7, 8]. The dynamic behavior of Mpro exemplifies its possibility to become a striking target for drug design. Moreover, SARS-CoV-2 Mpro is not similar to human homologous proteases [9]. The ligand-binding site of Mpro is placed into the groove of domains I and II comprising the crucial catalytic dyad His 41 and Cys 145 [10, 11]. Moreover, some recent studies demonstrated that Mpro could be the prominent target of SARS-CoV-2 infection [11-14].
Still today, there is no specific anti-SARS-CoV-2 drugs are available. However, several clinical trials are underway; and most of them are focused on relieving the symptoms [15]. Besides, the antiviral efficiency of several already existing drugs has been testified in several studies [16, 17]. However, repurposed drugs have proven effective, but their efficacy and safety are still ambiguous [18-20]. Besides, the recent coronavirus strain (B.1.351) found in South Africa possesses more infection rate, and it shows the ability to re-infect people. Recently, South Africa [21] and several European countries, including Austria, Estonia, Iceland, Italy, Lithuania, Luxembourg, Latvia, and Norway, has postponed the use of the AstraZeneca vaccine following reports of blood clots [22]. Nevertheless, the benefits outweigh the rare blood clot events, and the European Medical Agency, World Health Organization, and the International Society on Thrombosis and Hemostasis recommended taking the vaccine. However, it raises the urgency to find more specific drugs to inhibit SARS-CoV-2 infection with broader efficacy to overcome public concern.
Plant-derived compounds could be a great source of antiviral drug compounds as they possess low toxicity, have a more convenient biosynthesis process, and can be screened easily through computational biology techniques. Besides, most drug candidates used from the last four decades were derived from natural sources [23-25]. Also, plant synthesized compounds have shown antiviral activity against several viruses’ including’s Chikungunya [26], SARS [27], and SARS-CoV-2 [28].
This study was conducted to find out the potent natural anti-SARS-CoV-2 compounds from Syzygium aromaticum (clove). Syzygium aromaticum is commonly used as a spice, which contains many bioactive compounds and is cultivated worldwide [29, 30]. S. aromaticum has also been used as a traditional medicine for a long time [30]. Besides, S. aromaticum compounds have been shown to act against many viruses such as Hepatitis C virus, Herpes simplex virus [30], Feline calicivirus [31], and Adenovirus [32]. The potential antiviral activity of S. aromaticum against several RNA viruses raises the possibility to act against SARS-CoV-2. Thus, this study was projected to find out probable compounds against SARS-CoV-2 targeting Mpro.
METHODS AND MATERIALS
Isolation and preparation of ligands
In this study, initially, we built a compound dataset of S. aromaticum through related literature search on Scopus, Google Scholar, PubMed, and Web of science literature repository [33]. We curated 20 compounds of S. aromaticum from these databases and downloaded their three-dimensional structure from the Pubchem database [34] The PyRx ligand preparation wizard was used to prepare compounds as ligands (Version Python prescription 0.8) [35] through Merck molecular force field (mmff94) [36], and the ligands were then converted into PDBQT format for further analysis.
Preparation of receptor
The Mpro 3D structure (PDB ID: 6LU7) [37] was extracted from the largest crystal structure repository, Protein Data Bank (https://www.rcsb.org/) [38]. Before molecular docking, the receptor was prepared using Chimera [39] and AutoDock tools integrated into PyRx [35].
ADMET analysis
The physicochemical properties of isolated ligands were evaluated by ADMET analysis. ADMET profiling analysis is a promising and cost-reductive approach that tells us about any compound’s physicochemical properties, drug-likeness properties, potentiality, and effectiveness [40]. In silico studies have accelerated the velocity of drug design and are now widely used in pharmaceuticals, leading to finding novel compounds to combat various microorganisms [41]. Lipinski’s rule of five is essential for determining a drug’s probability with a particular pharmacological and biological activity [42]. Three or more violations do not follow the drug-likeness requirements and are not considered a drug for further study. ADMET properties were analyzed using the Schrodinger QikProp (QikProp, Schrödinger, LLC, New York, NY, USA) program [43]. The drug-likeness properties of the selected compounds were studied using Lipinski’s “rule of five” [44].
Compound’s screening
The virtual screening was conducted using AutoDock wizard integrated [35, 45] PyRx software (Version Python prescription 0.8) [35]. The ligands were kept as flexible, and the receptor was inflexible. The docking grid box (x = -13.09, y = 15.00, z = 69.32) was generated using Auto Grid engine in PyRx. The conformational root-mean-square deviation (RMSD) result of less than 1.0 Å was taken as perfect and bunched for later promising binding analysis. The highest negative score was considered as better binding. Here, α-ketoamide was considered as a control ligand [10]. The BIOVIA Visualizer (Discovery Studio v 4.5) was employed to observe molecular interactions [46].
Molecular dynamics simulation
The molecular dynamics simulation was conducted using the “WebGRO for Macromolecular Simulations (https://simlab.uams.edu/)” server utilizing the “GROMACS” macromolecular simulation system [47]. Initially, the ligand topology files were prepared by the “PRODRG” server [48]. In this study, the GROMOS96 43a1 force field was utilized, along with the SPC water model and NaCl (0.15 M) solvated cubic box. The energy was minimized using the steepest descent algorithm (5000 steps). For temperature control, NVT/NPT temperature (300 K) system was used in 1 bar pressure. Finally, we conducted a 50 ns simulation. The trajectory was used to calculate RMSD, Rg (Radius of gyration), RMSF (Root mean square fluctuation), SASA (Solvent accessible surface area), and Hydrogen bond analysis.
RESULTS
ADMET analysis
The QikProp ADME/Tox analysis protocol deciphered that all compounds follow “rule of 5” except bicornin (shown in Table 1). According to drug-likeness property analysis, the selected compounds’ molecular weights were between the recommended range (≤500 g/mol) except for bicornin. The hydrogen bond acceptor and donor were also below the recommended range (≤10 and ≤5, respectively). The filtered 19 compounds were then employed for further investigation.
Table 1. ADMET properties of all compounds.
Compound’s library screening and interaction visualization
The binding affinity of all selected ligands is shown in Table 2. The top graded anti-Mpro hits were selected based on their interaction with the catalytic dyad His41 and Cys145 and higher binding affinity. We found three compounds, i.e., oleanolic acid, crategolic acid, and kaempferol having higher binding affinity -7.7 kcal/mol, -7.6 kcal/mol, and -7.6 kcal/mol, respectively. Besides, they have shown crucial molecular interactions; thereby chosen for further analysis.
The molecular interaction analysis showed that all the selected compounds either interact with Cys145 and His41 or, at least with one of them. α-ketoamide is a positive control in this study that forms four H bonds with Gln189 residue and several alkyl bonds with Cys145, Met49, Met165, Leu27 & His41 residues (Figure 1a). Crategolic acid comprises H bond with Thr25, Leu141, and Gly143 residues and pi-alkyl bonds with Cys145, His41, Met165, and Met49 residues (Figure 1b). Oleanolic acid comprises H-bond with Ser144 and pi-alkyl bonds with Cys145 and Met49 residues (Figure 1c). The last compound, kaempferol, comprises H-bond with His163, pi-alkyl bonds with Cys145 and Met165, and pi-stacked bond with His41 residue (Figure 1d).
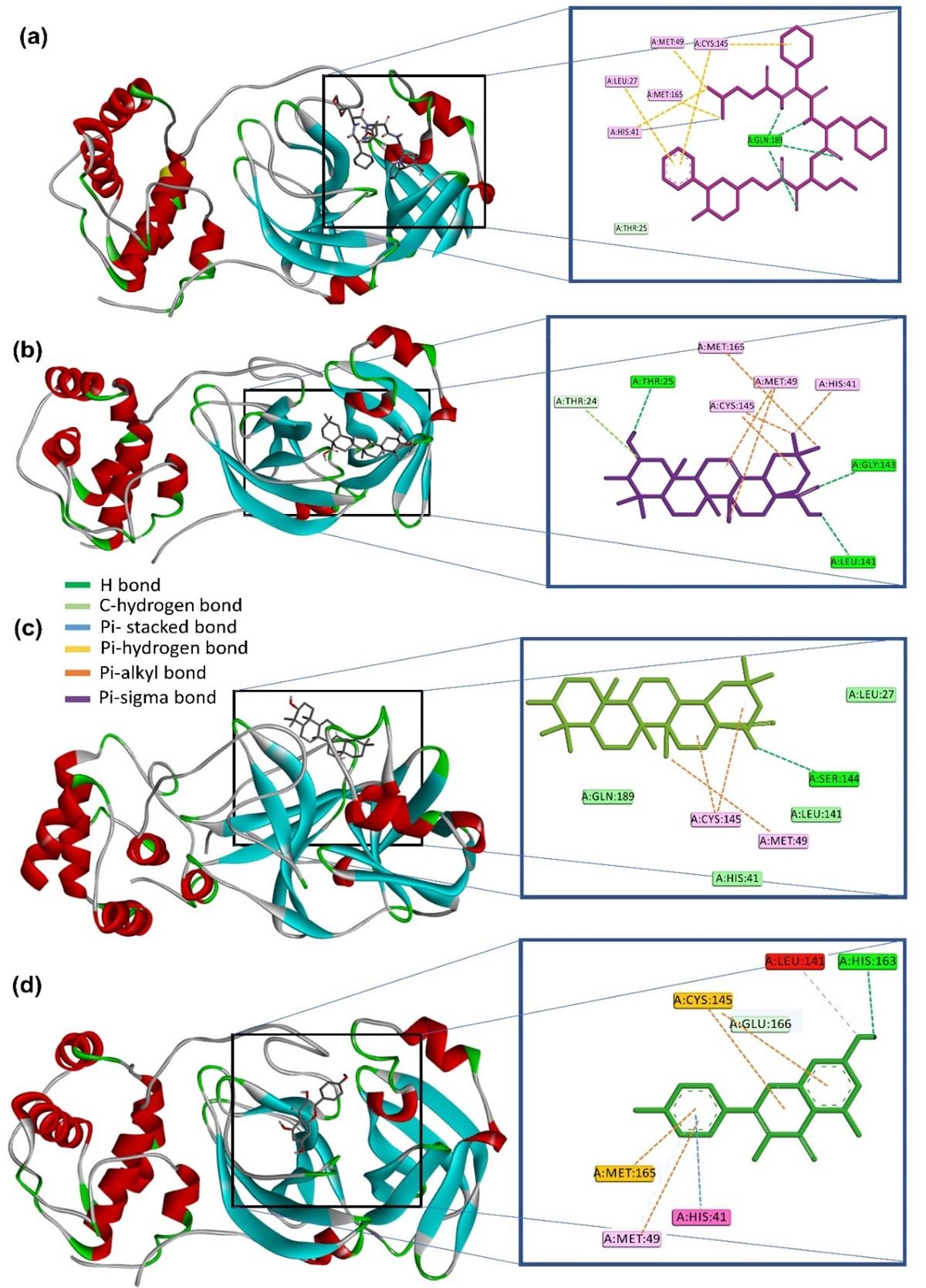
Table 2. Binding affinity of Syzygium aromaticum compounds with Mpro
Molecular dynamics simulation
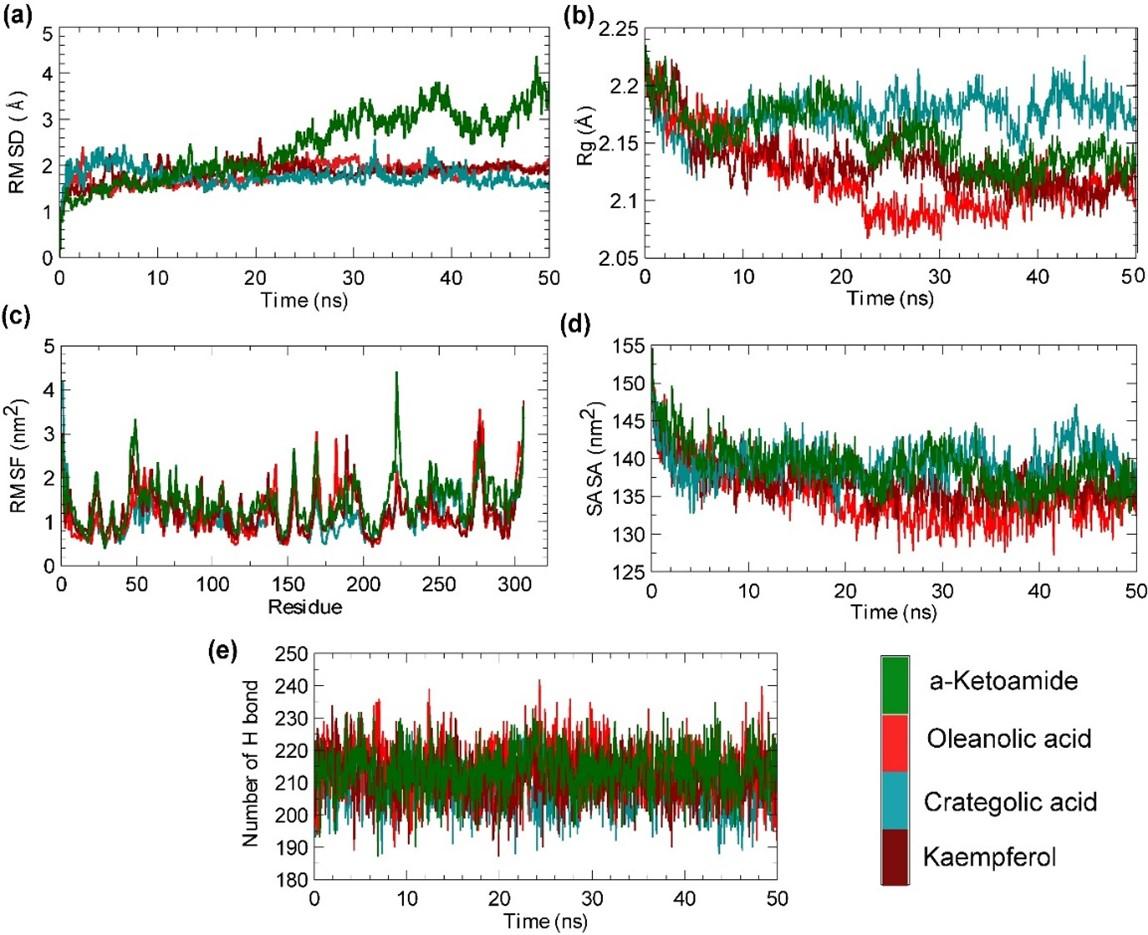
Molecular dynamics simulation was used to project the behaviour of projected compounds in the biological system. In this study, both control complex and newly selected compounds’ behaviors were studied through RMSD, Rg, RMSF, SASA and H bond studies. In Figure 2a, it is seen that the selected compounds have lower RMSD values than the positive control α-ketoamide. Crategolic acid showed relatively lower RMSD values than other compounds. The average RMSD values of α-ketoamide, crategolic acid, oleanolic acid, and kaemferol were 2.07 Å, 1.51 Å, 1.55 Å, and 1.57 Å, respectively. Interestingly, crategolic acid, oleanolic acid, and kaempferol showed a more stable condition in simulation compared to the control. The radius of gyration demonstrates the compactness of the system over time. The average Rg values of crategolic acid, oleanolic acid, and kaempferol were 2.17 Å, 2.12 Å, and 2.13 Å, respectively (Figure 2b). Oleanolic acid and kaempferol have lower Rg values compared to the control 2.15 Å. The fluctuation pattern of each amino acid residue was calculated using RMSF (Figure 2c). Figure 2c showed notable fluctuations in the terminal regions for each complex, but fewer fluctuations were seen in the active site region. The average RMSF values of α-ketoamide, crategolic acid, oleanolic acid, and kaemferol were 1.25 nm2, 0.97 nm2, 0.97 nm2, and 1.04 nm2, respectively. The solvent-accessible surface area was also evaluated for each complex. The higher SASA value demonstrates the openness of the systems. The average SASA values of the selected complex were 139.02 nm2, 139.45 nm2, 135.05 nm2, and 136.80 nm2 for α-ketoamide, crategolic acid, oleanolic acid, and kaempferol, respectively (Figure 2d). The positive control and crategolic acid have almost similar SASA values, and interestingly oleanolic acid and kaemferol have lower SASA values than control. The intermolecular hydrogen bonds play a vital role in deciphering the proper functions of any small molecules with receptors. Thus, we also calculated the hydrogen bonds of our system (Figure 2e). The oleanolic acid has more hydrogen bonds than α-ketoamide, while the rest two compounds have relatively similar hydrogen bonds. The number of hydrogen bonds also in parallel with other calculations, which depicts the compactness of our simulated systems.
DISCUSSION
This research aimed to identify potential SARS-CoV-2 Mpro drug candidates from natural sources [37]. Mpro has been investigated as an effective target to restrain the expansion of SARS-CoV-2 contamination. We have considered Syzygium aromaticum because it contains several bioactive compounds [49, 50]. In addition, it was found to have antioxidant activity and a broad range of pharmacological efficiency [51, 52]. Besides, Syzygium aromaticum has traditional history to use as common spice around the world. Peoples consumes Syzygium aromaticum daily.
It has been shown that α-ketoamide interacts with the residues of Mpro, namely His41, Gly143, Ser144, Cys145, His163, His164, Glu166, Pto168, and Gln189 [53]. Our study also found that α-ketoamide interacts with almost similar residues that rectify our methods for further study (shown in Figure 2b). Besides, the N3 (native ligand) of the chosen main protease (6LU7) interacts with His41 and Cys145 residues [11], which implies that His41 and Cys145 residues are crucial for SARS-CoV-2 Mpro inhibition. Moreover, recent studies showed that the phytochemicals form strong interactions with Leu27, His41, Met49, Cys145, Met165, Thr190 residues of SARS-CoV-2 Mpro [45, 54, 55]. In addition to the main protease, phytochemicals showed antiviral activity against SARS-CoV-2 envelope protein [56].
Among the studied 20 compounds of Syzygium aromaticum, only four compounds were used for more investigation considering their binding affinity and compared to the known antagonist α-ketoamide. All these compounds form hydrogen or hydrophobic interactions with the crucial residues His41-Cys145 of Mpro. In a previous study, oleanolic acid was described as an active compound against the hepatitis C virus (HCV) [57]. Likewise, Kaempferol was suggested to be an excellent anti-coronavirus candidate [58]. Recently, Jo et al. showed the anti-SARS-CoV-2 activity of kaempferol [59]. Khan et al. also showed that kaempferol bonds with the essential active site residue, inhibiting SARS-CoV-2 [60]. Moreover, kaempferol was also shown in several other pharmacological activities [61]. Besides, the presented compounds (Table 1) showed considerable bio-activities in different in-vitro studies. For example, recently, Alhadrami et al. depicted that olive-derived phytochemicals inhibit SARS-CoV-2 main protease at IC50 = 3.22–14.55 µM [62]. Furthermore, Colunga Biancatelli and colleagues reported the possible synergistic benefits of quercetin and vit-C against COVID-19 [63]. In addition, a clinical study denoted that quercetin improves the patient’s condition [64].
Our study found that all of our selected compounds follow Lipinski’s rule of five except bicornin. The selected three compounds, crategolic acid, oleanolic acids, and kaempferol, showed considerable water solubility, whereas bicornin failed to fulfill these parameters also. Also, these compounds showed considerable in vitro hERG toxicity. However, the only bicornin violates the Lipinski rule of five (shown in Table 1).
Molecular dynamics simulation is an effective technique to understand the stability and dynamics of the protein-ligand complex [65, 66]. The lower RMSD and RMSF values indicate the higher stability of the complex [66, 67]. The compounds, crategolic acid, oleanolic acid, and kaemferol formed stable complex with SARS-CoV-2 Mpro, though crategolic acid showed a sudden surge initially, but overall, it showed stable binding. Nukoolkarn et al. (2008) conducted two ns simulations and found that inhibitor compound binds with His41 and Cys145 residues of SARS-CoV 3CLpro [68]. Besides, several recent molecular simulation studies showed similar results [45, 67, 69]. Similarly, our identified compounds crategolic acid, oleanolic acid, and kaemferol also interacted with the active side residues His41 and Cys145 and formed stable conformation, which depicts their possible effectiveness over time.
CONCLUSION
The highly infectious nature of SARS-CoV-2 has possessed a devastating effect on human life all around the world. Therefore, SARS-CoV-2 antagonists are desperately needed to reduce the fast transmissibility of the virus. The major goal of this research was to find novel inhibitors for the SARS-CoV-2 main protease. This study employed several computational approaches to identify the probable antagonist of SARS-CoV-2 Mpro from 20 bioactive phytochemicals of Syzygium aromaticum. Considering the outputs of ADMET analysis, molecular docking, and molecular dynamics simulation, three compounds, crategolic acid, oleanolic acids, and kaempferol, showed satisfactory results to inhibit SARS-CoV-2 infection targeting the main protease (Figure 3). The identified compounds can be considered as lead molecules to develop drugs against COVID-19. However, more studies are required to confirm their activity and efficacy.
ACKNOWLEDGEMENTS
None
AUTHOR’S CONTRIBUTION
MAHMJ, and MCA conceived the plan of this research. MCA and MSR run the experiment. MCA, AJN, RAH, RAR, MAM, and MSK wrote the manuscript and analyzed the data. MCA prepared the final draft. MAHMJ, RAH, MSR, MKA, MAM, and MMR edited the manuscript. All authors revised and approved the manuscript for final submission.
CONFLICTS OF INTEREST
There is no conflict of interest among the authors.
References
- [1]Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet. 2020;395:497-506.
- [2]Farabi S, Ranjan Saha N, Anika Khan N, Hasanuzzaman M. Prediction of SARS-CoV-2 Main Protease Inhibitors from Several Medicinal Plant Compounds by Drug Repurposing and Molecular Docking Approach. ChemRxiv. 2020.
- [3]Benvenuto D, Giovanetti M, Ciccozzi A, Spoto S, Angeletti S, Ciccozzi M. The 2019-new coronavirus epidemic: Evidence for virus evolution. Journal of medical virology. 2020;92:455-9.
- [4]ul Qamar MT, Alqahtani SM, Alamri MA, Chen L-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. Journal of pharmaceutical analysis. 2020;10:313-9.
- [5]Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Hu Y, et al. Complete genome characterisation of a novel coronavirus associated with severe human respiratory disease in Wuhan, China. BioRxiv. 2020.
- [6]Tahir ul Qamar M, Alqahtani SM, Alamri MA, Chen L-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. Journal of Pharmaceutical Analysis. 2020;10:313-9.
- [7]Xue X, Yu H, Yang H, Xue F, Wu Z, Shen W, et al. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. Journal of virology. 2008;82:2515-27.
- [8]Wang F, Chen C, Tan W, Yang K, Yang H. Structure of main protease from human coronavirus NL63: insights for wide spectrum anti-coronavirus drug design. Scientific reports. 2016;6:1-12.
- [9]Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y, Jung SH. An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. Journal of medicinal chemistry. 2016;59:6595-628.
- [10]Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409-12.
- [11]Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, et al. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020:1-5.
- [12]Das S, Sarmah S, Lyndem S, Singha Roy A. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. Journal of biomolecular structure & dynamics. 2020:1-11.
- [13]Gentile D, Patamia V, Scala A, Sciortino MT, Piperno A, Rescifina A. Putative Inhibitors of SARS-CoV-2 Main Protease from A Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study. Marine drugs. 2020;18.
- [14]Bacha U, Barrila J, Velazquez-Campoy A, Leavitt SA, Freire E. Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry. 2004;43:4906-12.
- [15]Zhu R-f, Gao R-l, Robert S-H, Gao J-p, Yang S-g, Zhu C. Systematic Review of the Registered Clinical Trials of Coronavirus Diseases 2019 (COVID-19). medRxiv. 2020.
- [16]Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell research. 2020;30:269-71.
- [17]Colson P, Rolain JM, Lagier JC, Brouqui P, Raoult D. Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. International journal of antimicrobial agents. 2020;55:105932.
- [18]Magagnoli J, Narendran S, Pereira F, Cummings T, Hardin JW, Sutton SS, et al. Outcomes of hydroxychloroquine usage in United States veterans hospitalized with Covid-19. medRxiv. 2020.
- [19]McGonagle D, O’Donnell JS, Sharif K, Emery P, Bridgewood C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. The Lancet Rheumatology. 2020.
- [20]Mehra MR, Desai SS, Ruschitzka F, Patel AN. RETRACTED: Hydroxychloroquine or chloroquine with or without a macrolide for treatment of COVID-19: a multinational registry analysis. Lancet (London, England). 2020.
- [21]Welle D. COVID: Several European countries halt use of AstraZeneca vaccine (Accessed on March 12, 2021). https://wwwdwcom/en/covid-several-european-countries-halt-use-of-astrazeneca-vaccine/a-56835406. 2021.
- [22]Wise J. Covid-19: European countries suspend use of Oxford-AstraZeneca vaccine after reports of blood clots. BMJ. 2021;372:n699.
- [23]Newman DJ, Cragg GM. Natural Products as Sources of New Drugs from 1981 to 2014. Journal of natural products. 2016;79:629-61.
- [24]Newman DJ, Cragg GM. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. Journal of Natural Products. 2020;83:770-803.
- [25]Atanasov AG, Zotchev SB, Dirsch VM, Orhan IE, Banach M, Rollinger JM, et al. Natural products in drug discovery: advances and opportunities. Nature Reviews Drug Discovery. 2021;20:200-16.
- [26]Oo A, Rausalu K, Merits A, Higgs S, Vanlandingham D, Bakar SA, et al. Deciphering the potential of baicalin as an antiviral agent for Chikungunya virus infection. Antiviral research. 2018;150:101-11.
- [27]Chen F, Chan KH, Jiang Y, Kao RY, Lu HT, Fan KW, et al. In vitro susceptibility of 10 clinical isolates of SARS coronavirus to selected antiviral compounds. Journal of clinical virology : the official publication of the Pan American Society for Clinical Virology. 2004;31:69-75.
- [28]Liu H, Ye F, Sun Q, Liang H, Li C, Li S, et al. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro. Journal of enzyme inhibition and medicinal chemistry. 2021;36:497-503.
- [29]Lazreg Aref H, Gaaliche B, Fekih A, Mars M, Aouni M, Pierre Chaumon J, et al. In vitro cytotoxic and antiviral activities of Ficus carica latex extracts. Natural product research. 2011;25:310-9.
- [30]Batiha GE, Alkazmi LM, Wasef LG, Beshbishy AM, Nadwa EH, Rashwan EK. Syzygium aromaticum L. (Myrtaceae): Traditional Uses, Bioactive Chemical Constituents, Pharmacological and Toxicological Activities. Biomolecules. 2020;10.
- [31]Aboubakr HA, Nauertz A, Luong NT, Agrawal S, El-Sohaimy SA, Youssef MM, et al. In vitro antiviral activity of clove and ginger aqueous extracts against feline calicivirus, a surrogate for human norovirus. Journal of food protection. 2016;79:1001-12.
- [32]Moradi M-T, Karimi A, Alidadi S, Hashemi L. Anti-adenovirus activity, antioxidant potential, and phenolic content of dried flower buds of Syzygium aromaticum extract in HEp2 cell line. Marmara Pharma J. 2017;21:852-9.
- [33]Ali M, Munni YA, Das R, Akter N, Das K, Mitra S, et al. In silico chemical profiling and identification of neuromodulators from Curcuma amada targeting Acetylcholinesterase. Network Modeling Analysis in Health Informatics and Bioinformatics. 2021;10:1-16.
- [34]Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem 2019 update: improved access to chemical data. Nucleic acids research. 2019;47:D1102-d9.
- [35]Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Chemical biology: Springer; 2015. p. 243-50.
- [36]Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. Journal of Computational Chemistry. 1996;17:490-519.
- [37]Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289-93.
- [38]Rose PW, Prlić A, Altunkaya A, Bi C, Bradley AR, Christie CH, et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic acids research. 2016:gkw1000.
- [39]Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25:1605-12.
- [40]Talele TT, Khedkar SA, Rigby AC. Successful applications of computer aided drug discovery: moving drugs from concept to the clinic. Current topics in medicinal chemistry. 2010;10:127-41.
- [41]Ivanov AS, Veselovsky AV, Dubanov AV, Skvortsov VS. Bioinformatics platform development: from gene to lead compound. Methods in molecular biology (Clifton, NJ). 2006;316:389-431.
- [42]Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced drug delivery reviews. 1997;23:3-25.
- [43]Ligprep M, Macromodel G. QikProp; Schrodinger, LLC; New York, NY, 2011.
- [44]Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced drug delivery reviews. 2001;46:3-26.
- [45]Ali MC, Nur AJ, Khatun MS, Dash R, Rahman MM, Karim MM. Identification of potential SARS-CoV-2 main protease inhibitors from Ficus Carica latex: An in-silico approach. J Adv Biotechnol Exp Ther. 2020;3:57-67.
- [46]Biovia DS. Discovery studio visualizer v4.5. Release; 2015.
- [47]Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19-25.
- [48]Schüttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta crystallographica Section D, Biological crystallography. 2004;60:1355-63.
- [49]Diego C-rF, Wanderley OP. Clove (Syzygium aromaticum): a precious spice. Asian Pacific Journal of Tropical Biomedicine. 2014:90-6.
- [50]Shan B, Cai YZ, Sun M, Corke H. Antioxidant capacity of 26 spice extracts and characterization of their phenolic constituents. Journal of agricultural and food chemistry. 2005;53:7749-59.
- [51]Nassar MI, Gaara AH, El-Ghorab AH, Farrag A, Shen H, Huq E, et al. Chemical constituents of clove (Syzygium aromaticum, Fam. Myrtaceae) and their antioxidant activity. Revista Latinoamericana de Química. 2007;35:47.
- [52]Salim B, Said G, Noureddine M, Hocine A, Angelika BA. A note study on antidiabetic effect of main molecules contained in clove using molecular modeling interactions with DPP-4 enzyme. International Journal of Computational and Theoretical Chemistry. 2017;5:9.
- [53]Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell research. 2020;30:269-71.
- [54]Narkhede RR, Pise AV, Cheke RS, Shinde SD. Recognition of Natural Products as Potential Inhibitors of COVID-19 Main Protease (Mpro): In-Silico Evidences. Natural products and bioprospecting. 2020;10:297-306.
- [55]Kumar Y, Singh H, Patel CN. In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. Journal of infection and public health. 2020;13:1210-23.
- [56]Orfali R, Rateb ME, Hassan HM, Alonazi M, Gomaa MR, Mahrous N, et al. Sinapic Acid Suppresses SARS CoV-2 Replication by Targeting Its Envelope Protein. Antibiotics. 2021;10.
- [57]Kong L, Li S, Liao Q, Zhang Y, Sun R, Zhu X, et al. Oleanolic acid and ursolic acid: Novel hepatitis C virus antivirals that inhibit NS5B activity. Antiviral Research. 2013;98:44-53.
- [58]Schwarz S, Sauter D, Wang K, Zhang R, Sun B, Karioti A, et al. Kaempferol derivatives as antiviral drugs against the 3a channel protein of coronavirus. Planta medica. 2014;80:177.
- [59]Jo S, Kim S, Shin DH, Kim MS. Inhibition of SARS-CoV 3CL protease by flavonoids. Journal of enzyme inhibition and medicinal chemistry. 2020;35:145-51.
- [60]Khan A, Heng W, Wang Y, Qiu J, Wei X, Peng S, et al. In silico and in vitro evaluation of kaempferol as a potential inhibitor of the SARS-CoV-2 main protease (3CLpro). Phytotherapy research : PTR. 2021;35:2841-5.
- [61]Wang L, Tu Y-C, Lian T-W, Hung J-T, Yen J-H, Wu M-J. Distinctive antioxidant and antiinflammatory effects of flavonols. Journal of Agricultural and Food Chemistry. 2006;54:9798-804.
- [62]Alhadrami HA, Sayed AM, Sharif AM, Azhar EI, Rateb ME. Olive-Derived Triterpenes Suppress SARS COV-2 Main Protease: A Promising Scaffold for Future Therapeutics. Molecules. 2021;26.
- [63]Colunga Biancatelli RML, Berrill M, Catravas JD, Marik PE. Quercetin and Vitamin C: An Experimental, Synergistic Therapy for the Prevention and Treatment of SARS-CoV-2 Related Disease (COVID-19). Frontiers in immunology. 2020;11:1451.
- [64]Di Pierro F, Iqtadar S, Khan A, Ullah Mumtaz S, Masud Chaudhry M, Bertuccioli A, et al. Potential Clinical Benefits of Quercetin in the Early Stage of COVID-19: Results of a Second, Pilot, Randomized, Controlled and Open-Label Clinical Trial. International journal of general medicine. 2021;14:2807-16.
- [65]Peele KA, Potla Durthi C, Srihansa T, Krupanidhi S, Ayyagari VS, Babu DJ, et al. Molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: A computational study. Informatics in Medicine Unlocked. 2020;19:100345.
- [66]Munni YA, Ali MC, Selsi NJ, Sultana M, Hossen M, Bipasha TH, et al. Molecular simulation studies to reveal the binding mechanisms of shikonin derivatives inhibiting VEGFR-2 Kinase. Computational Biology and Chemistry. 2020:107414.
- [67]ClinicalTrials.gov. Phase I Clinical Trial in Healthy Adult (PICTHA) (https://clinicaltrials.gov/ct2/show/NCT04313127) (Accessed on 22 march 2020). 2020.
- [68]Nukoolkarn V, Lee VS, Malaisree M, Aruksakulwong O, Hannongbua S. Molecular dynamic simulations analysis of ritronavir and lopinavir as SARS-CoV 3CLpro inhibitors. Journal of theoretical biology. 2008;254:861-7.
- [69]Mahmud S, Uddin MAR, Zaman M, Sujon KM, Rahman ME, Shehab MN, et al. Molecular docking and dynamics study of natural compound for potential inhibition of main protease of SARS-CoV-2. Journal of Biomolecular Structure and Dynamics. 2020:1-9.