Human immunodeficiency virus type 1: Role of proteins in the context of viral life cycle
Abstract
The Acquired Immunodeficiency Syndrome (AIDS) is a major global pandemic and of paramount public health concern. Over the years, antiretroviral therapy (ART) has emerged as the gold standard of AIDS treatment. However, drug resistance and toxicity, and drug accessibility and compliance issues have blunted its positive impacts. Thirty years into the AIDS pandemic, there is a critical need of the hour to identify and develop an effective treatment strategy that can be successfully implemented with high levels of coverage. The HIV-1 life cycle bestows numerous potential targets for therapeutic intervention, however, only a few have been exploited till date. HIV- 1 encodes fifteen viral proteins from just nine genes. The expression of structural polyproteins Gag, Pol, and Env enable the assembly of the virion, the regulatory proteins Rev and Tat regulate viral gene expression and the accessory proteins Vpu, Nef, Vpr, and Vif equip the virion to evade or counteract host immune defenses. In this review article, we highlight each structural, regulatory, and accessory protein’s role in the context of the HIV-1 life cycle. We also discuss anti-retroviral therapy, its advantages, and shortcomings. Finally, we provide insight into various unexploited and potential therapeutic targets in the life cycle of HIV
Keywords
INTRODUCTION
The current AIDS pandemic is a complex combination of diverse sub-epidemics within and between different countries of the world and can undoubtedly be termed a public-health crisis. The first known case of AIDs was reported in 1981. Since then, the Joint United Nations Program on HIV/AIDS (UNAIDS) has estimated that AIDS has been the cause of demise for more than 32 million people worldwide and an estimated 37.9 million people were living with AIDS by the end of 2018. The Human Immunodeficiency Virus (HIV) is the etiological agent of AIDS. Two types of HIV virus have been isolated and characterized from infected patients: HIV-1 and HIV-2. HIV-1 was discovered first. It is the most found strain of the virus and is more prevalent worldwide. Whereas HIV-2 is mainly restricted to Western Africa [1]. The aim of this review article is to emphasize the role of each structural, regulatory, and accessory protein with respect to the HIV-1 life cycle leading to HIV infection. Further, we have discussed about the highly preferred treatment therapy for AIDS that is anti-retroviral therapy and its benefits as well as drawbacks. In addition to this, an attempt has been made to explore the possible therapeutic targets which can be exploited for drug and vaccine development.
THE HUMAN IMMUNODEFICIENCY VIRUS
Virion structure
HIV-1 is a spherical retrovirus and has a diameter of ~ 100–120 nm. The virion has an outer envelope, which is a lipid bilayer and embedded in it are glycoproteins gp120 and gp41. At the centre of the virion lies a cone-shaped core, which is composed of the viral capsid protein (p24). Enclosed within the core is the genetic material comprising of two positive-sense single-stranded linear RNA molecules ((+) ssRNA) of length ~ 10kilobase (kb). Tightly bound to the RNA is nucleocapsid protein (p7). Besides (+) ssRNA, the core also encapsulates two molecules of the enzyme reverse transcriptase (p64), a protease (p10), and an integrase (p32). Between the inner viral capsid core and outer envelope lies a layer of the matrix protein (p17) [2].
Tropism
HIV-1, like other viruses, is an obligate intracellular parasite. Humans are the natural hosts of HIV-1. The virus can infect disparate cells of the immune system such as CD4+ T cells, dendritic cells, and macrophages. However, it predominantly infects and kills CD4+T cells. This leads to a depletion in the CD4+ T cell population, resulting in profound immunodeficiency, thus incapacitating the immune system, and impairing the patient’s ability to fight opportunistic infections. The main mechanism for CD4+ T cell depletion is apoptosis i.e., programmed cell death which can be induced by multiple pathways. This decrease in the number of CD4+ cells in a patient’s body indicates the development of HIV-1 infection [3].
Genome organization
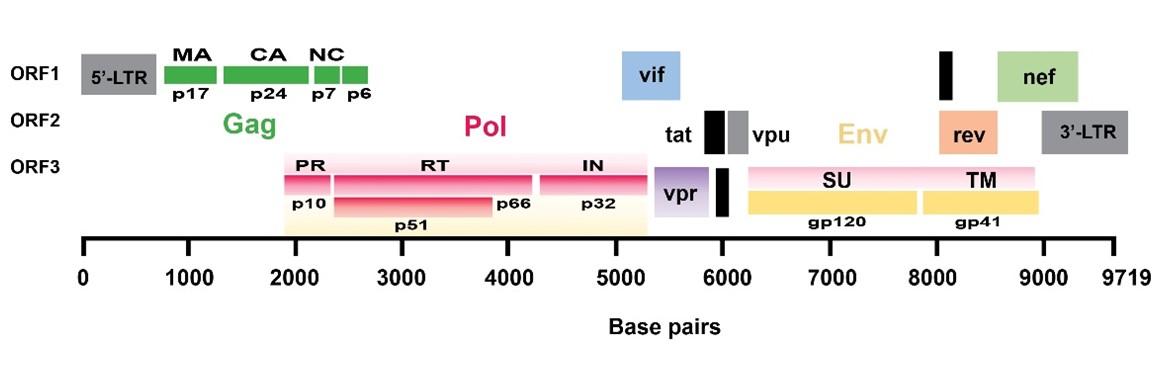
The HIV-1 genome has three fundamental defining open-reading frames (ORFs) (Figure 1). The Gag gene is one of these three prime ORFs. It encodes the polyprotein Gag (group-specific antigen). Gag is named p55, based on its molecular weight of 55 kD. It is initially synthesized as a precursor protein, that during or shortly after virus budding, is cleaved by the viral protease (PR) into mainly three cleavage products – matrix protein (p17), capsid protein (p24), and nucleocapsid protein (p7) [4].
The second ORF in HIV-1, pol, encodes Gag-Pol (p160) polyprotein containing the precursor to the enzymes protease (PR), reverse transcriptase (RT), and integrase (IN). The third ORF, env, encodes the viral protein Env, a glycoprotein of 160 kD (aptly named gp160). After translation, gp160 is cleaved into the surface (SU) glycoprotein gp120 and the transmembrane (TM) glycoprotein gp41. Together they interact with the target receptor (CD4) and co-receptor (CXCR4 or CCR5) on host cells [1].
In addition to the retroviral ORFs gag, pol, and env, HIV-1 has six supplementary ORFs that encode two regulatory and four axillary proteins. The HIV-1 regulatory proteins Rev (regulator of virion expression) and Tat (trans activator of transcription) gene are expressed early in the virus’s life cycle. The replication of HIV-1 virion in host cells is intimately linked and completely reliant upon numerous cellular host factors. Conversely, host cells also harbour intrinsic resistance factors (also called restriction factors) that suppress viral replication [5]. The array of axillary proteins encoded by HIV distinguishes it from other retroviruses. They’re called axillary because they don’t have any enzymatic or structural capabilities but aid the virion in evading and manipulating the host’s innate and adaptive immune responses. Vpu, Vif, Nef, and Vpr are the four axillary proteins encoded by HIV-1 [6]. The host-HIV-1 interactome can help fully understand how HIV-1 gains control of the host and how the cell attempts to counteract infection by elucidating these critical interactions between the virion and the host protein.
PROTEINS INVOLVED IN HIV-1 LIFE CYCLE
The HIV-1 life cycle can be arbitrarily divided into two distinct phases: the early stage, which refers to the steps where the virion binds and enters into the target cell, conversion of viral RNA to cDNA (reverse transcription) occurs, delivery of cDNA to the site of integration and integration of the viral cDNA into the cell genome takes place, and the late phase, which includes the expression of the HIV gene products and the assembly, release, and maturation of progeny virion [7] (Figure 2).
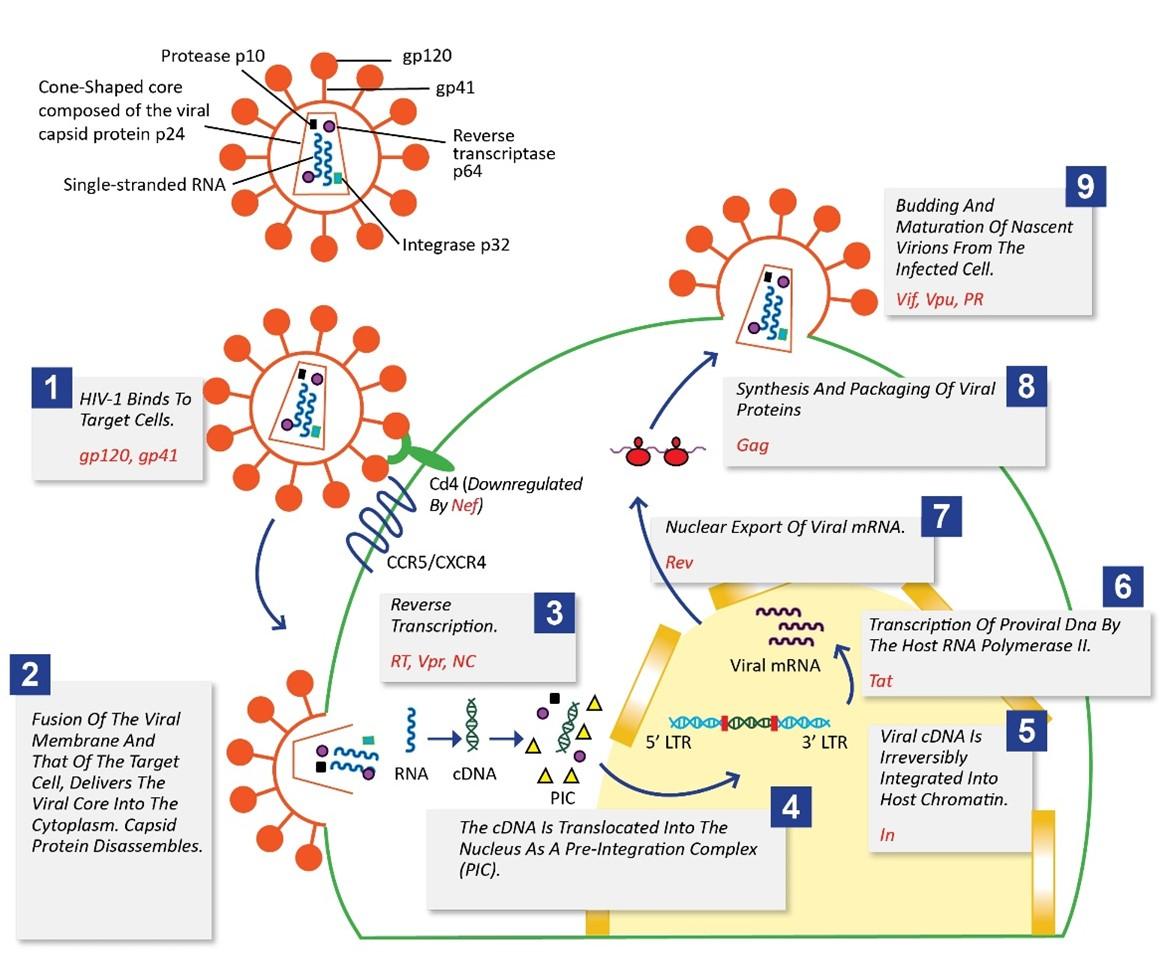
Proteins involved in binding and entry of the virus into the target cells
The envelope forms the outermost layer of the virion and is a lipid bilayer ∼4 nm thick, typically containing ∼300,000 lipids. It is derived from the host’s cell membrane. Identification and characterization of the array of proteins in enveloped viruses like HIV-1 crucial as these proteins often direct infection. These proteins are the only antigenic determinants on intact virion that are visible to the immune system and hence, are considered as primary targets for vaccine development [8-9].
Interspersed and embedded in the HIV-1 viral envelope are virally encoded glycoproteins – the surface glycoprotein gp120 and the transmembrane glycoprotein gp41. The two are non-covalently linked to each other and form a single subunit of a trimeric “spike” on the surface of the virion. Projecting from the surface of each virion are 72 such glycoprotein spikes, each composed of gp120 and gp41 [10]. When HIV-1 infects target cells, it employs its surface glycoproteins to adhere and fuse to the host cell membrane. HIV-1 entry is a multiphasic process that is initiated when gp120 selectively binds to the primary receptor CD4. This binding triggers a conformational change in gp120 and augments its affinity for a secondary receptor, or “co-receptor” either C-X-C chemokine receptor type 4 (CXCR4) or C-C chemokine receptor type 5 (CCR5) [11]. It was found that individuals possessing naturally occurring 32-base pair deletion in the CCR5 gene are highly resistant to infection by primary HIV-1 isolates which further confirmed the above-mentioned phenomenon. These individuals are seemingly healthy, suggesting the absence of a functional CCR5 will not have unexpected side-effects and is not associated with any significant immunological dysfunction [12]. Thus, the seven-transmembrane G protein-coupled chemokine co-receptor CCR5 and CXCR4 are seen as a potential therapeutic target. Maraviroc belongs to a class of anti-retroviral drugs called CCR5 antagonists that exhibits potent anti-human immunodeficiency virus type 1 (HIV-1) activity as well as favorable pharmacological properties. Maraviroc selectively binds to CCR5 subsequently blocking the interaction of the surface (SU) glycoprotein gp120 with it. Thus, preventing the membrane fusion event which are required for viral entry. According to the previous studies, it was seen that Maraviroc does not affect CCR5 cell surface levels or associated intracellular signaling, indicating that it a functional antagonist of CCR5 [13][14,15]. Following gp120’s attachment to the coreceptor, subsequent conformational changes in both gp120 and gp41 cause a fusion of viral and cellular membranes, delivering the viral core into the target cell’s cytoplasm and allowing reverse transcription to begin [16]. Enfuvirtide is a synthetic peptide that blocks HIV-1 entry into host cells by interfering with virus-cell fusion by competitively binding to gp41 and blocking the formation of the post-fusion structure. It is the first and currently the only fusion inhibitor approved by the US Food and Drug Administration (FDA) which is used in combination with other antiretroviral agents [17]. Despite the importance of the primary receptor CD4 during viral entry, it is well known that HIV-1 suppresses the homologous CD4 receptor’s cell surface expression. CD4 downregulation is thought to give the virus a selective replicative advantage in vivo. Increased CD4 expression on the cell surface has been shown to interfere with viral particle release and infectivity in studies. The presence of CD4 during the virus life cycle causes issues such as superinfection, early CD4 binding to nascent virus particles, and virus release inhibition. Thus, the removal of cell-surface CD4 is required for optimal virus production by HIV-1 [18]. Negative factor (Nef) is 27–35 kD myristoylated protein. Myristoylation is the covalent attachment of myristic acid to the N terminus glycine residue. Nef is highly expressed in the early stages of the viral replication cycle. Nef mediates CD4 down-regulation by inducing endocytosis of CD4 followed by its degradation through the endo-lysosomal pathway [19].
Proteins involved in uncoating and reverse transcription
After the fusion of the viral membrane and that of the target cell, the capsid protein disassembles, and the viral RNA enters the cytoplasm. This is followed by reverse transcription, i.e., synthesis of linear double-stranded cDNA from an RNA template and is catalyzed by the 64kD enzyme reverse transcriptase. It is an asymmetric heterodimer consisting of two subunits – the larger p66 subunit and the smaller p51 subunit. Reverse transcription is a hallmark of the retroviral replication cycle [20]. Virus populations have extensive genetic heterogeneity due to the mutation-prone nature of viral replication and are referred to as quasi-species. The human immunodeficiency virus is one of the most polymorphic viruses known and is an important and prominent example of a viral quasi-species. Patients infected with HIV-1 harbor a highly diverse virus population with many different mutants. Spontaneous Mutations are generated since reverse transcriptase (RT) lacks proofreading mechanisms and thus has low fidelity. It is this high-level genetic diversity that allows HIV-1 to escape the immune system, and rapidly evolve drug resistance, making it difficult to come up with effective vaccination strategies and drug therapy [21]. The viral protein R (Vpr) is a small (14 kD) accessory protein, that is well conserved in HIV-1. Vpr is multifunctional and plays various roles at multiple stages of the HIV-1 viral life cycle. It modulates the in vivo mutation rate of HIV-1 by influencing the accuracy of the reverse transcription, thus playing a key role in the regulation of HIV-1[22].
Proteins involved in the viral integration
The cDNA interacts with viral and cellular proteins and is translocated into the nucleus as a nucleoprotein pre-integration complex (PIC). The PIC is a large complex consisting of cDNA, integrase, reverse transcriptase, MA protein, CA protein, and Vpr protein. Due to its large size, it cannot passively diffuse across the nuclear envelope. Vpr expression is said to induce transient bulges in the nuclear envelope, which sometimes burst, creating a channel between the nucleus and the cytoplasm, thus facilitating the nuclear import of the pre-integration complex into the nucleus[22]. The cDNA is irreversibly integrated into host chromatin by the 32 kD enzyme integrase. Integration can essentially occur at any location in the genome, however certain regions of chromatin are preferred. This integrated viral DNA is known as proviral DNA. Blocking integration is of unique importance, as once integrated, identification and destruction of the hidden provirus is nearly impossible. Raltegravir is the first approved integrase inhibitor used for treatment of AIDs and has rapid, potent and sustained effects. Integration occurs in two steps, namely 3′ end processing, and DNA strand transfer. Raltegravir interferes with the strand transfer reaction, and this prevents the incorporation of linear HIV-1 cDNA into the host cell’s genome and disrupts the viral life cycle of HIV-1 [23-24].
Proteins involved in gene expression
The proviral DNA, like a cellular gene, is transcribed by the host RNA polymerase II (RNAPII). Both ends of the proviral DNA are flanked by a repeat sequence known as the long terminal repeat (LTR). LTR is the control centre for gene expression, as it contains multiple upstream DNA regulatory elements that serve as binding sites and interact with a myriad of cellular transcription initiation factors. The 5′ LTR region codes for the promotor for transcription of the viral genes [25]. Tat (transactivator of transcription) is a 14-kD nuclear transcription factor encoded by the tat gene in HIV-1. Tat is an RNA binding protein that enhances transcription by recognizing and binding to a cis-acting RNA sequence called TAR element (transactivator response element), present within the extended terminal repeat region at the 5′ end of the nascent viral mRNA transcript [26-27].
Nascent Viral RNAs undergo processing in the nucleus, just like cellular mRNAs, that include the addition of a 5′ methylguanine ‘cap’ and a 3′ polyA tail. HIV-1 primary transcripts fall into 3 classes according to their degree of splicing: (i) 9.2 kb unspliced transcripts, that encodes for the Gag and Pol polyproteins or can be packaged into virions to serve as genomic RNA; (ii) 4 kb singly spliced transcripts coding for the viral proteins Env, Vpu, Vif and Vpr; (iii) 2 kb multiply spliced transcripts that express Tat, Rev and Nef [28]. Unlike cellular mRNAs that are usually spliced to completion before being exported to the cytoplasm, a significant fraction of retroviral transcripts remains unspliced but are still exported to the cytoplasm. Rev is a small (16kD) trans-acting regulatory protein that shuttles between the nucleus and the cytoplasm. It binds to the Rev response element (RRE), that is present on unspliced and singly spliced viral RNAs transcripts and exports these RNAs from the nucleus to the cytoplasm [29-30]. The HIV-1 mRNA encodes three structural polyproteins Gag, Pol, and Env. Two of these polyprotein precursors Gag and Pol, as mentioned before, are derived from the same 9.2 kb unspliced transcript. The gag gene encodes matrix proteins (MA), capsid protein (CA), and nucleocapsid protein (NC) in the 5′ to 3′ direction of the reading frame (NC). Protein p6, spacer peptide 1 (SP1) between CA and NC, and spacer peptide 2 (SP2) between NC and p6 are likewise found in HIV-1 Gag. HIV-1 protein synthesis is also entirely dependent on the translation machinery of the host cell and takes place on cellular ribosomes. About the gag coding region, the pol coding region of the mRNA now partially overlaps and is in the 1 reading frame. As a result, when ribosomes translating gag reach the start of the pol coding area, they must slip back one nucleotide to exit the gag reading frame. A Gag-Pol fusion protein is produced due to a single ribosomal frameshift. In general, Gag is produced by 95% of translating ribosomes, while a Gag-Pol fusion protein is produced by 5%. The precursor proteins are not generated with similar stoichiometry, despite the fact that they are derived from the same mRNA [31].
Proteins involved in the assembly and budding of nascent virions from the infected cells
One significant way of understanding the molecular mechanisms underlying the assembly of HIV-1 virion is by studying assembly in a defined system in vitro. The expression of Gag has been found to be is central to HIV-1 assembly, as in the absence of other viral components, it alone can spontaneously assemble into spherical immature virus-like particles (VLPs) in vitro, which cannot be morphologically distinguished via an electron microscope from authentic immature (non-infectious) virus [32].
Understanding the mechanism of virus assembly and release has been hampered by the resolution limitations of traditional microscopy. The immature virus-like particles are roughly spherical, with a diameter of 100–150 nm, and contain 5000 rod-shaped Gag molecules tightly packed side-by-side in a radial fashion within the immature particles, with the N-terminal MA domain associated with the membrane and the C-terminal portion of Gag oriented toward the particle’s centre, according to high-resolution cryogenic electron microscopy analyses [33]. Gag can multimerize and produce virus-like particles on its own, but to make infectious virions, it must interact with various viral and host components during the assembly process [33]. Gag is a multi-domain viral protein, as previously stated. Each domain of Gag is involved in the assembly and release process, and it is made up of three independently folded conserved structural domains. HIV-1 Gag takes part in Gag-membrane, Gag-Gag, and Gag-RNA interactions. The Gag-membrane contact is mediated by Gag’s N-terminal MA domain, which directs Gag to the host cell’s plasma membrane, where it binds Gag to the lipid bilayer’s inner leaflet, which is the primary site of Gag multimerization and particle formation. The CA domain of the Gag polyprotein is required for Gag-Gag interactions. Also, CA domain along with the NC domain, homo oligomerizes to facilitate Gag multimerization and particle assembly. The Gag-RNA interaction is promoted by a particular interaction between Gag’s NC domain and viral genome sequences. The NC domain binds to viral genomic RNA, causing two copies of the viral genomic RNA to be packaged into each virus particle. The viral RNA genome transmits all genetic information to new host cells, including the instructions needed to produce new virions, and is thus critical for virion infectivity [34-35].
In recent years, significant progress has been made in understanding the structure and cell biology of Gag, revealing a plethora of prospective Gag-related therapeutic targets. Targeting gag gene products successfully blocks HIV-1 replication, providing a promising method for new classes of medicines to supplement existing HIV-1 therapy options [36]. Gag and Gag-Pol are targeted and trafficked to the assembly site, which in most cases is the plasma membrane, by the 17 kD matrix (MA) domain. It also plays an essential role in the incorporation of the viral Env glycoproteins spikes during virus assembly. The N-terminal attachment of a myristic acid moiety is required for the MA domain’s membrane-targeting activity. We know this because a mutation in the N-terminal Gly residue, which is the site of myristic acid attachment, prevents Gag from binding to the membrane and prevents virus assembly. Gag molecules must first recognize a location in the plasma membrane where they can oligomerize into higher order multimers in order to start particle assembly [37]. Gag is found in specific PM microdomains called lipid rafts, which are dense and ordered groups of tightly packed saturated lipids stabilized by cholesterol, according to studies. Lipid rafts have a distinct molecular makeup than the surrounding membrane [38]. In the past, it was thought that HIV-1 assembly and effective particle release took place at the plasma membrane of all host cells. Several results and recently proposed models, however, suggest that HIV-1 assembly in primary macrophages is triggered by internal sequestration of Gag in late endosomal membranes. Later secretory pathways based on endosomes carry the formed virion to the plasma membrane. This is believed to control virion release in terms of time and space [36-37].
Protease (PR) is a dimeric virus-encoded proteolytic enzyme belonging to the family of aspartic proteases. During or shortly after budding PR performs a series of 12 sequential, highly specific, perfectly timed cleavage reactions on the Gag polyprotein precursor, producing the mature matrix protein p17 (MA), capsid protein p24 (CA), nucleocapsid protein p7 (NC), and protein p6 [39]. While the MA protein remains associated with the inner face of the virion membrane, the CA protein condenses and forms a characteristic fullerene shaped cone around the viral RNA. This rearrangement is known as virion maturation, and it produces a morphological transition from an immature to an infectious virion [40]. Due to its sensitive and essential function, HIV-1 protease has been widely exploited as a drug target and it exhibits a broad substrate recognition. The HIV-1 capsid is a conical protein shell made up of a hexameric lattice of the CA protein that repeats itself. In both the early and late stages of the viral replication cycle, the capsid is crucial. The capsid core is released into the cytoplasm shortly after the viral and host cell membranes fuse, where it disassembles via a process known as uncoating. Capsid stability is required for efficient reverse transcription and infectivity, according to site-directed mutagenesis investigations of HIV-1 variants. Capsid assembly and maturation are necessary to produce infectious virus particles throughout the late stages of the HIV-1 life cycle. The HIV-1 CA protein is composed of two individually folded domains: a 150 amino acid N-terminal domain (CANTD) and an 80 amino acid C-terminal domain, according to structural analyses (CACTD). A flexible linker connects the CANTD to the CACTD. The CACTD is orientated towards the interior of the HIV-1 capsid core, whereas the CANTD is positioned on the outside surface [41][42]. The capsid protein, which is a structural component of HIV-1, is responsible for the shape of the immature Gag and the mature viral core, which has a conical form. The discovery of multiple capsid inhibitors in recent years has emphasized the viral capsid’s importance as a promising target for therapeutic development. CAP-1 and CAP-2 were the first small compounds to be identified as HIV-1 capsid inhibitors. CAP-1 has been proven to inhibit HIV-1 replication by up to 95% while CAP-2 is lethal to cells [43]. CA is one of the most conserved proteins among HIV-1 subtypes, and mutations are not well tolerated. CA is a very appealing target for inhibitor development because of these properties [44].
The HIV-1 nucleocapsid (NC) proteins are small, basic, highly conserved, and multifunctional nucleic acid binding proteins. The nucleocapsid protein, is a highly conserved zinc finger protein. It plays a central role in virus replication, mainly by interacting with nucleic acids. About 2000 NC molecules bind to the viral RNA through electrostatic interactions between their basic residues and the phosphodiester backbone of RNA. Thus, NC coats the viral RNA and protects it from degradation by RNAases. NC also promotes the initiation and the strand transfer reactions, during reverse transcription. This is because NC acts as a nucleic acid chaperone and enables the rearrangement of the nucleic acid structure into most thermodynamically stable conformations by catalyzing the breakage and reformation of base pairs[45][46]. Novel therapeutic compounds for the treatment of AIDs must contend with the obstacle of drug resistance. Because they are mutationally intolerant and essential for both acute infection and virion assembly, the HIV-1 nucleocapsid protein is a particularly desirable antiviral target. The fact that point mutations of the NC protein result in non-infectious viruses is another convincing reason to explore NC as a promising therapeutic target [47].
The targeting of discrete steps in the HIV-1 replication cycle, such as Gag trafficking to the site of virus assembly, Gag binding to cellular membranes, Gag monomer interactions leading to multimerisation, and budding and release of nascent virions from the infected cell, provides a better strategy for new classes of drugs to complement existing HIV-1 treatment options [48]. Virion infectivity factor (Vif) is a phosphorylated protein with a size of 23 kD. The host apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (APOBEC3G; A3G) is encapsulated by budding virion in Virion lacking a functioning Vif gene. APOBEC3G is a DNA-editing enzyme that catalyses the conversion of cytosine (dC) to uracil (dU) in single-stranded DNA during reverse transcription, limiting HIV-1 replication and conferring intrinsic immunity to the host. HIV-1 averts the potent antiviral effects of the intrinsic resistance factor APOBEC3G by encoding Vif at relatively high levels late in the viral replication cycle. Vif forms a complex with APOBEC3G, thus preventing its packaging into assembling virions. This interaction between Vif and APOBEC3G is an attractive therapeutic target [41-42].
Viral protein U (Vpu) is the smallest (16 kD) of all HIV-1 proteins, having 81 amino acids and consisting of a single transmembrane domain. It is an accessory protein that is expressed by HIV-1 but not by HIV-2. NMR spectroscopy shows that it consists of two major domains: a short N-terminal hydrophobic transmembrane anchor domain (TMD), and a longer cytoplasmic domain (CD) [43]. One of the chief functions of Vpu is to aid in the release of progeny virus from infected target cells. The host restriction factor, bone marrow stromal cell antigen 2 (BST-2) is an interferon-induced transmembrane protein that tethers enveloped viruses such as HIV-1 to the cell surface, thus inhibiting their release [49]. Vpu relieves this restriction imposed by BST-2 by removing it from sites of virion assembly and budding at the plasma membrane and thus facilitates the virion release from infected cells. Since there isn’t any Vpu targeting drug currently available that has cleared clinical trials further than phase two, Vpu is a potential therapeutic target [43].
UPDATES ON THE TREATMENT OF HIV
AIDS was once thought of as “a very big threat” as it was feared to result in a “medical apocalypse” and was hence termed as a “curse upon the human race”. Ever since the HIV-1 virus was first discovered, tremendous progress has been made. Today anti-retroviral therapy has emerged as the cornerstone of HIV-1 treatment. It consists of using a cocktail or a combination of anti-retroviral drugs each of which targets and inhibits a step of the HIV-1 life cycle. ART maximally suppresses viral replication and reduces the plasma HIV-1 viral load (VL) i.e., the number of HIV-1 RNA copies per milliliter of blood, to levels that most clinical assays cannot detect (<50 RNA copies/mL) [50].
ART is however not curative and cannot eradicate HIV-1 infection completely, as viral reservoirs remain in the form of silent HIV-1 DNA integrated in the host genome of long-lived resting memory CD4 +T cells and are thus recognized by the immune system or eradicated by drugs. ART can, however, stop the progression of AIDS in patients. The availability and rapid scale-up of anti-retroviral therapy have transformed AIDs from an inevitably fatal disease to a chronic and controllable condition. Even though a significant decline in the death of patients due to AIDS is seen since the use of ART, global efforts have not corroborated to be ample, and the global prevalence of HIV has expanded since its discovery as the virus continues to spread with an alarming and increasing speed [51].
Drug resistance is a significant barrier that often restricts the efficacy of antiretroviral drugs that are already accessible. When drug levels aren’t high enough to stop viral reproduction, drug resistance develops. For the treatment of AIDS, there are now 25 FDA-approved anti-retroviral medications available, which can be divided into five categories. A mutation that provides resistance to one medicine now frequently confers resistance to other drugs in the same class. As a result, the establishment of drug resistance limits the number of drugs that can be used [47].
HIV-1 reverse transcriptase (RT), which converts single-stranded viral RNA into corresponding double-stranded proviral DNA, was the focus of early efforts to stop HIV-1 replication. Because RT is required for retrovirus replication but not found in HIV-1-susceptible host cells, it seemed an ideal target for selective therapeutic action. Anti-RT medications are divided into two categories: nucleoside RT inhibitors (NRTIs) and non-nucleoside RT inhibitors (NNRTIs) [52]. In addition to FDA-approved protease inhibitors (PIs) such as tipranavir, the sole FDA-approved fusion inhibitor for the treatment of AIDS is enfuvirtide. Among the FDA-approved integrase inhibitors are Raltegravir (RAL), Elvitegravir (EVG), and Dolutegravir (DTG). A medication regimen known as Highly Active Anti-Retroviral Therapy (HAART) containing a combination of NRTIs, NNRTIs, PIs, and entry inhibitors is used to treat HIV. In HIV patients, HAART has considerably reduced morbidity and mortality rates. Currently, increased emphasis is placed on including two, three, or even more medications that target the HIV life cycle [53].
A primary goal of current National Institute of Allergy and Infectious Diseases (NIAID) -funded HIV therapy research is to produce long-acting treatments that, unlike current antiretrovirals, may be given less often. Long-acting medicines, broadly neutralizing antibodies, and therapeutic vaccinations are the three categories of agents being investigated. The efficacy of two investigational long-acting HIV drugs, rilpivirine LA and cabotegravir LA, among people who have had trouble adhering to standard antiretroviral therapy was evaluated by NIAID. Another study included a combination of monthly cabotegravir LA injections and monthly infusions of VRC01LS, a broadly neutralizing antibody discovered by the NIAID, can keep HIV suppressed in people who have previously been treated with antiretroviral therapy [54]. HIV infections can be treated in three ways by broadly neutralizing antibodies: By attaching directly to the virus, it is prevented from entering a cell and its eradication is accelerated. Through attaching to an HIV-infected cell, immune-system components that assist cell death are recruited. Lastly, by means of creating a compound with a crucial piece of HIV, it may stimulate immune cells in a similar way to a vaccine, so preparing the immune system for future encounters with the virus. A therapeutic vaccination, unlike one designed to prevent HIV infection, would be administered to persons who are already infected with the virus. Such a vaccine would train the immune system to be ready for any future outbreak of HIV, obviating the need for additional treatment and maybe avoiding the requirement for booster shots. Such a strategy could lead to long-term viral remission, or treatment or vaccination that results in HIV levels that are undetectable for lengthy periods of time without the need for antiretroviral therapy [55].
CONCLUSION
This review article accentuates the life cycle of HIV-1 and the key role played by various viral proteins at different stages. One prime attribute of the virus, responsible for its evolutionary success is its ‘deceptive simplicity’, i.e., even though HIV-1 has a small genome, consisting of just 9 genes that encode for only 15 mature proteins, the virus is difficult to target. ART is unfolding to abate the drug burden but also exhibits detrimental side effects. Moreover, ART solely cannot cure or exterminate HIV. Subsequently, insightful expansion to novel therapies and trailblazing paradigms should be exploited. It is requisite to recognize other factors that are imperative for viral replication in the host cell. Meticulous probing of complexities cognate to HIV infection, role of immune cells and molecular alterations during the infection will assist in treatment. This would also aid in impeding the phenomenon of resistance developed by HIV. It is also crucial to determine the viral target cell and investigate the target cell dependent variations in viral replication cycle.
Despite advancement, many people living with HIV remain undiagnosed due to lack of awareness and prevention. Vanquishing this obstacle by diagnosing and treating the infection at a primary stage would assist in mitigating the proliferation of the virus. Therefore, exigency of collaborative scientific and social measures cannot be left unnoticed. Congruous variations in treatment regimens can aid patients subjugate the prospective impediments that come along with the disease. Factors like pervasiveness of psychiatric disorders, socioeconomic status, meagre health literacy and social stigma are also reckoned as radical hurdles in treatment of this infection. Mental health plays a very crucial role in combating any illness, consequently, expedient counselling regarding treatment obedience and psychological consolation should be promulgated in patients and the society.
ACKNOWLEDGEMENT
None.
AUTHOR CONTRIBUTIONS
VJ and KR conceptualized this manuscript. SD, RP, MR, and NS aligned the literature, VJ, KG, KR and VD wrote the manuscript. NK, and SD prepared the figures. NS, MR, and KG did proof reading and corrections. All authors contributed to the revision and approved the final manuscript.
CONFLICTS OF INTEREST
There is no conflict of interest among the authors.
References
- [1]Seitz R. Human Immunodeficiency Virus (HIV). Transfus Med Hemotherapy 2016;43:203–22. https://doi.org/10.1159/000445852.
- [2]McLaren PJ, Fellay J. HIV-1 and human genetic variation. Nat Rev Genet 2021;22:645–57. https://doi.org/10.1038/s41576-021-00378-0.
- [3]Vijayan KV, Karthigeyan KP, Tripathi SP, Hanna LE. Pathophysiology of CD4+ T-Cell depletion in HIV-1 and HIV-2 infections. Front Immunol 2017;8:1–8. https://doi.org/10.3389/fimmu.2017.00580.
- [4]Klingler J, Anton H, Réal E, Zeiger M, Moog C, Mély Y, et al. How HIV-1 gag manipulates its host cell proteins: A focus on interactors of the nucleocapsid domain. Viruses 2020;12. https://doi.org/10.3390/v12080888.
- [5]Colomer-Lluch M, Ruiz A, Moris A, Prado JG. Restriction Factors: From Intrinsic Viral Restriction to Shaping Cellular Immunity Against HIV-1. Front Immunol 2018;9:1–18. https://doi.org/10.3389/fimmu.2018.02876.
- [6]Hernandez L. Accessory Regulatory Proteins of HIV-1 and Host Restriction Factors Interactions. Biomed J Sci Tech Res 2020;31:24308–12. https://doi.org/10.26717/bjstr.2020.31.005120.
- [7]Mailler E, Bernacchi S, Marquet R, Paillart JC, Vivet-Boudou V, Smyth RP. The life-cycle of the HIV-1 gag–RNA complex. Viruses 2016;8:1–19. https://doi.org/10.3390/v8090248.
- [8]Xiao T, Cai Y, Chen B. Hiv-1 entry and membrane fusion inhibitors. Viruses 2021;13:1–19. https://doi.org/10.3390/v13050735.
- [9]Burnie J, Guzzo C. The incorporation of host proteins into the external HIV-1 envelope. Viruses 2019;11:1–25. https://doi.org/10.3390/v11010085.
- [10]Kumar R, Ozorowski G, Kumar V, Holden LG, Shrivastava T, Patil S, et al. Characterization of a stable HIV-1 B/C recombinant, soluble, and trimeric envelope glycoprotein (Env) highly resistant to CD4-induced conformational changes. J Biol Chem 2017;292:15849–58. https://doi.org/10.1074/jbc.M117.803056.
- [11]Shaik MM, Peng H, Lu J, Rits-Volloch S, Xu C, Liao M, et al. Structural basis of coreceptor recognition by HIV-1 envelope spike. Nature 2019;565:318–23. https://doi.org/10.1038/s41586-018-0804-9.
- [12]Hütter G, Neumann M, Nowak D, Klein S, Klüter H, Hofmann WK. The effect of the CCR5-delta32 deletion on global gene expression considering immune response and inflammation. J Inflamm 2011;8:1–8. https://doi.org/10.1186/1476-9255-8-29.
- [13]Wilen CB, Tilton JC, Doms RW. HIV: Cell binding and entry. Cold Spring Harb Perspect Med 2012;2. https://doi.org/10.1101/cshperspect.a006866.
- [14]Sayana S, Khanlou H. Maraviroc: a new CCR5 antagonist. Http://DxDoiOrg/101586/14787210719 2014;7:9–19. https://doi.org/10.1586/14787210.7.1.9.
- [15]Lin H-Y, Ho Y, Liu H-L. Structure-Based Pharmacophore Modeling to Discover Novel CCR5 Inhibitors for HIV-1/Cancers Therapy. J Biomed Sci Eng 2019;12:10–30. https://doi.org/10.4236/jbise.2019.121002.
- [16]El-Zohairy MA, Zlotos DP, Berger MR, Adwan HH, Mandour YM. Discovery of Novel CCR5 Ligands as Anticolorectal Cancer Agents by Sequential Virtual Screening. ACS Omega 2021;6:10921–35. https://doi.org/10.1021/acsomega.1c00681.
- [17]Lai YT. Small molecule hiv-1 attachment inhibitors: Discovery, mode of action and structural basis of inhibition. Viruses 2021;13:1–12. https://doi.org/10.3390/v13050843.
- [18]Liu Q, Zhang P, Lusso P. Quaternary interaction of the hiv-1 envelope trimer with cd4 and neutralizing antibodies. Viruses 2021;13. https://doi.org/10.3390/v13071405.
- [19]Kwon Y, Kaake RM, Echeverria I, Suarez M, Karimian Shamsabadi M, Stoneham C, et al. Structural basis of CD4 downregulation by HIV-1 Nef. Nat Struct Mol Biol 2020;27:822–8. https://doi.org/10.1038/s41594-020-0463-z.
- [20]Ha B, Larsen KP, Zhang J, Fu Z, Montabana E, Jackson LN, et al. High-resolution view of HIV-1 reverse transcriptase initiation complexes and inhibition by NNRTI drugs. Nat Commun 2021;12:1–11. https://doi.org/10.1038/s41467-021-22628-9.
- [21]Cuevas JM, Geller R, Garijo R, López-Aldeguer J, Sanjuán R. Extremely High Mutation Rate of HIV-1 In Vivo. PLoS Biol 2015;13:1–19. https://doi.org/10.1371/journal.pbio.1002251.
- [22]Byeon IJL, Calero G, Wu Y, Byeon CH, Jung J, DeLucia M, et al. Structure of HIV-1 Vpr in complex with the human nucleotide excision repair protein hHR23A. Nat Commun 2021;12. https://doi.org/10.1038/s41467-021-27009-w.
- [23]Anker M, Corales RB. Raltegravir (MK-0518): A novel integrase inhibitor for the treatment of HIV infection. Expert Opin Investig Drugs 2008;17:97–103. https://doi.org/10.1517/13543784.17.1.97.
- [24]Normale E, Cachan S De. R Altegravir : M Olecular B Asis of I Ts M Echanism of a Ction 2009;14:5–16.
- [25]Ne E, Palstra RJ, Mahmoudi T. Transcription: Insights From the HIV-1 Promoter. Int Rev Cell Mol Biol 2018;335:191–243. https://doi.org/10.1016/bs.ircmb.2017.07.011.
- [26]Ensoli B, Moretti S, Borsetti A, Maggiorella MT, Buttò S, Picconi O, et al. New insights into pathogenesis point to HIV-1 Tat as a key vaccine target. Arch Virol 2021;166:2955–74. https://doi.org/10.1007/s00705-021-05158-z.
- [27]Kamori D, Ueno T. HIV-1 tat and viral latency: What we can learn from naturally occurring sequence variations. Front Microbiol 2017;8:1–7. https://doi.org/10.3389/fmicb.2017.00080.
- [28]Leblanc J, Weil J, Beemon K. Posttranscriptional regulation of retroviral gene expression: Primary RNA transcripts play three roles as pre-mRNA, mRNA, and genomic RNA. Wiley Interdiscip Rev RNA 2013;4:567–80. https://doi.org/10.1002/wrna.1179.
- [29]Blissenbach M, Grewe B, Hoffmann B, Brandt S, Überla K. Nuclear RNA Export and Packaging Functions of HIV-1 Rev Revisited. J Virol 2010;84:6598–604. https://doi.org/10.1128/jvi.02264-09.
- [30]Pai S, Mudgal J, Kamath BV, Pai KSR. An insight on promising strategies hoping to cure HIV-1 infection by targeting Rev protein—short review. Pharmacol Reports 2021;73:1265–72. https://doi.org/10.1007/s43440-021-00257-9.
- [31]Hung M, Patel P, Davis S, Green SR. Importance of Ribosomal Frameshifting for Human Immunodeficiency Virus Type 1 Particle Assembly and Replication. J Virol 1998;72:4819–24. https://doi.org/10.1128/jvi.72.6.4819-4824.1998.
- [32]Scarlata S, Carter C. Role of HIV-1 Gag domains in viral assembly. Biochim Biophys Acta – Biomembr 2003;1614:62–72. https://doi.org/10.1016/S0005-2736(03)00163-9.
- [33]de Marco A, Müller B, Glass B, Riches JD, Kräusslich HG, Briggs JAG. Structural analysis of HIV-1 maturation using cryo- electron tomography. PLoS Pathog 2010;6. https://doi.org/10.1371/journal.ppat.1001215.
- [34]Dilley KA, Nikolaitchik OA, Galli A, Burdick RC, Levine L, Li K, et al. Interactions between HIV-1 Gag and Viral RNA Genome Enhance Virion Assembly. J Virol 2017;91. https://doi.org/10.1128/jvi.02319-16.
- [35]Sundquist WI, Kra H. HIV-1 Assembly, Budding, and Maturation 2012:1–24.
- [36]Welsch S, Keppler OT, Habermann A, Allespach I, Krijnse-Locker J, Kräusslich HG. HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog 2007;3. https://doi.org/10.1371/journal.ppat.0030036.
- [37]Pelchen-Matthews A, Kramer B, Marsh M. Infectious HIV-1 assembles in late endosomes in primary macrophages. J Cell Biol 2003;162:443–55. https://doi.org/10.1083/jcb.200304008.
- [38]Yamasaki. 基因的改变NIH Public Access. Bone 2014;23:1–7. https://doi.org/10.1016/j.virusres.2009.04.007.Lipids.
- [39]Yang H, Nkeze J, Zhao RY. Effects of HIV-1 protease on cellular functions and their potential applications in antiretroviral therapy. Cell Biosci 2012;2:1–8. https://doi.org/10.1186/2045-3701-2-32.
- [40]Sakuragi JI. Morphogenesis of the infectious HIV-1 virion. Front Microbiol 2011;2:1–5. https://doi.org/10.3389/fmicb.2011.00242.
- [41]Goila-Gaur R, Strebel K. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology 2008;5:1–16. https://doi.org/10.1186/1742-4690-5-51.
- [42]Britan-Rosich E, Nowarski R, Kotler M. Multifaceted counter-APOBEC3G mechanisms employed by HIV-1 Vif. J Mol Biol 2011;410:1065–76. https://doi.org/10.1016/J.JMB.2011.03.058.
- [43]Ruiz A, Guatelli JC, Stephens EB. The Vpu protein: new concepts in virus release and CD4 down-modulation. Curr HIV Res 2010;8:240–52. https://doi.org/10.2174/157016210791111124.
- [44]Carnes SK, Sheehan JH, Aiken C. Inhibitors of the HIV-1 capsid, a target of opportunity. Curr Opin HIV AIDS 2018;13:359–65. https://doi.org/10.1097/COH.0000000000000472.
- [45]Krishnamoorthy G, Roques B, Darlix JL, Mély Y. DNA condensation by the nucleocapsid protein of HIV-1: A mechnism ensuring DNA protection. Nucleic Acids Res 2003;31:5425–32. https://doi.org/10.1093/nar/gkg738.
- [46]Levin JG, Mitra M, Mascarenhas A, Musier-Forsyth K. Role of HIV-1 nucleocapsid protein in HIV-1 reverse transcription. RNA Biol 2010;7:754–74. https://doi.org/10.4161/rna.7.6.14115.
- [47]Arts EJ, Hazuda DJ. HIV-1 antiretroviral drug therapy. Cold Spring Harb Perspect Med 2012;2. https://doi.org/10.1101/cshperspect.a007161.
- [48]Waheed AA, Freed EO. HIV type 1 Gag as a target for antiviral therapy. AIDS Res Hum Retroviruses 2012;28:54–75. https://doi.org/10.1089/aid.2011.0230.
- [49]Evans DT, Serra-Moreno R, Singh RK, Guatelli JC. BST-2/tetherin: a new component of the innate immune response to enveloped viruses. Trends Microbiol 2010;18:388–96. https://doi.org/10.1016/J.TIM.2010.06.010.
- [50]Deeks SG, Archin N, Cannon P, Collins S, Jones RB, de Jong MAWP, et al. Research priorities for an HIV cure: International AIDS Society Global Scientific Strategy 2021. Nat Med 2021;27:2085–98. https://doi.org/10.1038/s41591-021-01590-5.
- [51]Henrich TJ, Hatano H, Bacon O, Hogan LE, Rutishauser R, Hill A, et al. HIV-1 persistence following extre277277mely early initiation of antiretroviral therapy (ART) during acute HIV-1 infection: An observational study. PLoS Med 2017;14:1–22. https://doi.org/10.1371/journal.pmed.1002417.
- [52]De Clercq E, Li G. Approved antiviral drugs over the past 50 years. Clin Microbiol Rev 2016;29:695–747. https://doi.org/10.1128/CMR.00102-15.
- [53]Singh, AnuradhaSingh, VishalSingh RK. Recent Advances in Treatment of HIV/AIDS Infection and its Social Implication. Res Gate 2020.
- [54]Antiretroviral Drug Discovery and Development | NIH: National Institute of Allergy and Infectious Diseases n.d. https://www.niaid.nih.gov/diseases-conditions/antiretroviral-drug-development (accessed March 25, 2022).
- [55]Future Directions for HIV Treatment Research | NIH: National Institute of Allergy and Infectious Diseases n.d. https://www.niaid.nih.gov/diseases-conditions/future-hiv-treatment (accessed March 25, 2022).