Developing a multiepitope vaccine against dengue virus in Bangladesh using immunoinformatics approach
Abstract
Dengue fever is a devastating mosquito-borne illness that has claimed the lives of countless people. The virus responsible for this disease is a member of the Flaviviridae family, which produces positive-stranded RNA. Dengue fever is an exquisite viral fever caused by the bite of an Aedes mosquito carrying one of four serotypes of dengue virus. This virus is transmitted via a vertical route utilizing a comprehensively unique system. Unfortunately, no effective vaccine has yet been developed to eradicate this disease. This study employed computational methods to design and propose a multi-epitope vaccine against dengue virus in Asia. This study utilized various immunoinformatics databases to predict potent epitopes on the envelope protein of the dengue virus using in silico methods. We identified a total of 14 epitopes from the target envelope protein by assessing their ability to induce both innate and acquired immunity through T- and B-lymphocyte-mediated responses. Because dengue virus is an RNA virus, epitope conservation was considered, and all selected epitopes were 100 percent conserved. The antigenicity of the final component of the multi-epitope vaccine was 0.7055. To improve the stability of the vaccine protein, disulfide engineering was performed in a region with high mobility. Additionally, codon adaptation and in silico cloning ensure that the proposed subunit vaccine will be expressed at a higher level in E. coli. In order to evaluate the binding free energy and stability of the combination, the vaccine protein and TLR-4 receptor were subjected to a molecular docking simulation. In order to establish active immunity against the dengue virus, the proposed in silico vaccine must be tested for safety and immunogenicity.
INTRODUCTION
Dengue is a serious disease transmitted by mosquitoes that has the potential to spread as a pandemic. Endemic dengue is a causative agent and it is a grave health warning for many developing tropical countries. They have been established in all tropical regions of the planet for more than six decades [1]. Southeast Asia is identified as the region with the highest prevalence of this disease in numerous tropical areas. The awe-inspiring hemorrhagic pattern of dengue fever has become the most dangerous cause of death in southeast Asia. This infection has also been documented in non-tropical regions of Asia, including East Asia and China [2]. Dengue occurs irregularly in Bangladesh, where the virus caused a widespread epidemic in 2000 and persisted until 1964. At that time, we discovered dengue in Bangladesh, where it was first identified, and identified factors beneficial to future dengue hemorrhagic fever epidemics [3]. Most likely, the outbreak started when a strain of the dengue virus came from a nearby country where it was common, and probably it was Thailand. In addition to the end of dichlorodiphenyltrichloroethane (DDT) spraying, the spread of disease was caused by weather, population, and lifestyle [4]. Even though it increased every other year, the highest number of cases was reported in 2002. Thus, we reduce the number of notifications that may be an artifact of the surveillance system [5]. Poll-based serological observation suggests that transmission of dengue is typical. Without intelligent interventions, future dengue risk will be exacerbated by unplanned urbanization, environmental decline, rising population mobility, and financial factors. Therefore, there is an urgent need to develop a vaccine for these high-risk regions. As part of this research, we need to use immunoinformatics to make a vaccine against the Asian-associated dengue virus that will work in Bangladesh. A flow chart discloses the full procedure of the study in Figure 1.
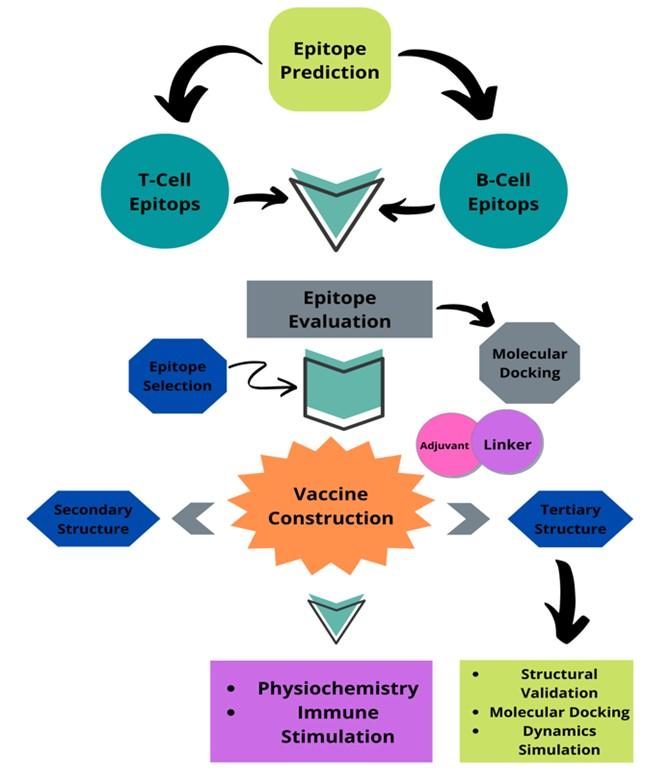
METHODS AND MATERIALS
Proteome salvation and antigen extracts
We selected probable HCMV proteomes from the viprbrc website database in order to pick antigens [6]. On the HCMV surface membrane, spike proteins were deposited. They collaborated with this host-binding protein to enter the human genome [7]. We measured the HCMV spike protein for the multiepitope vaccine strategy with the precise link between glycoproteins and disease. First, we selected the protein sequence of the dengue virus, which had been downloaded as a fasta file. The ddg-pharmfac database website was used to analyze the collected antigens having a threshold value of 0.4 [8]. Lastly, the spike protein with the highest antigenic score was chosen for further investigation.
Forecast and evaluation of helper T-lymphocyte epitopes
Helper T-cells (HTLs) are an essential component of adaptive immunity that recognizes different antigens and initiates B and cytotoxic T-cells, resulting in the destruction of the pathogen. Furthermore, B-cell and T-cell epitopes were predicted from dengue virus, as well as SARS coronavirus, which became disease and could be pandemic. To determine the HTL epitopes, we utilized the IEDB’s MHC class II essential allele prediction tool. The HTL epitopes were chosen using the Agreement method to support a percentile level of fifty [7]. These epitopes underwent additional testing and demonstrated antigenicity using Vaxijen server version 2.0 [9].
Forecast and evaluation of cytotoxic T-lymphocyte epitopes
Cytotoxic T lymphocytes are able to kill phagocytes via this mechanism directly [10]. Therefore, we utilized the NetCTL v1.2 server [11]. The collected epitopes were tested once more using the Vaxijen v2.0 [10], Toxiprod [13], and Allerrtop v2.0 [9] servers. All parameters are left at their defaults for all forecasts. A prediction threshold of 0.75 was established for CTL epitope identification [13].
Forecast and evaluation of linear B lymphocyte epitopes
B cell epitopes are needed to safely give humoral or antibody medication [14]. For this purpose, the online portal iBCE-EL (http://www.thegleelab.org/iBCE-EL/) utilized this with default levels [15].
Modeling of multi-epitope vaccine
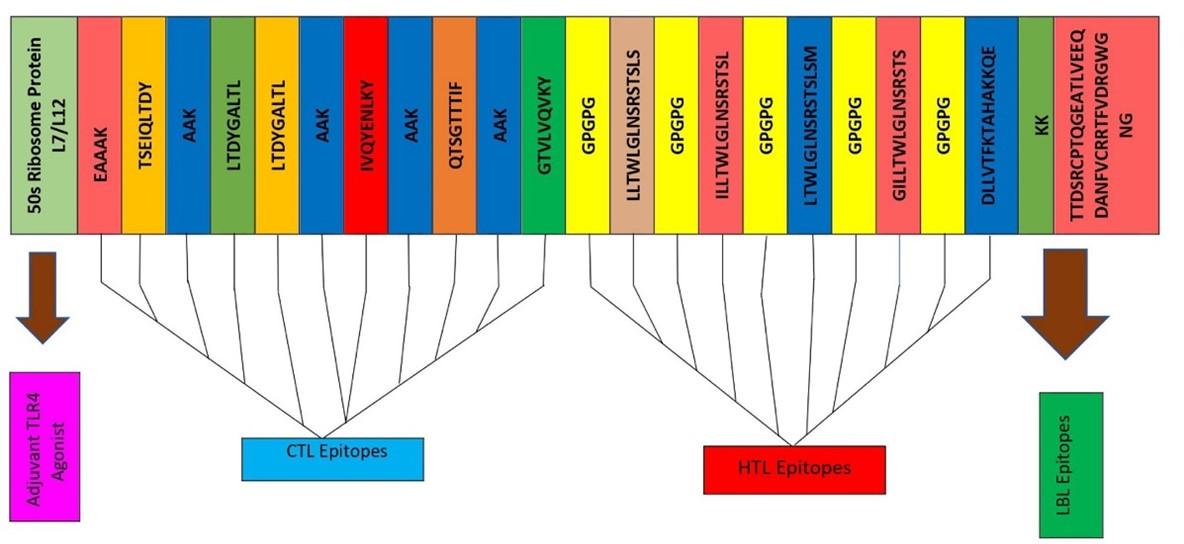
The vaccine was produced using the selected CTL epitope, HTL epitope, and LBL epitopes, a complete adjuvant, and the pertinent linkers [14; 16]. As an adjuvant for viral glycoprotein recognition, we used TLR4 agonist here [17; 18]. Therefore, 50S ribosomal protein (NCBI ID: P9WHE3) was valued as an adjuvant to enhance the immunogenicity of the candidate vaccine. The adjuvant was linked to the linker EAAAK. In contrast, the selected CTL was linked with (AAY) linkers, the HTL was linked with (GPGPG) linkers, and the LBL was linked with (KK) linkers [14; 16]. The AAY linker was utilized to affect protein equilibrium [19; 20]. The linkers effectively separate two epitopes to ensure that each epitope maintains its optimal function [13].
Physicochemical and immunological evaluation
The functional characteristics of the vaccine were predicted using the ProtParam database (http://web.expasy.org/protparam/) [21]. ProtParam is a program that calculates the various physical and chemical parameters of a protein sequence, such as its molecular weight, theoretical pI (isoelectric point), amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index, and grand average hydropathicity (GRAVY). Again, we used Vaxijen v2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) [22] to evaluate the vaccine’s immune properties by measuring its MHC-1 immunogenicity [9; 11] using Allertop, Biosoland, and SOLpro [8].
Secondary construction forecast
The SOPMA server (https://npsa-prabi.ibcp.fr/NPSA/npsa seccons.html) and PSIPRED v4.0 server (http://bioinf.cs.ucl.ac.uk/index.php?id=779) identified the vaccine model’s two-dimensional basic characteristics, such as alpha-helix and random coils, when given the vaccine model [23]. SOPMA produces a secondary structure prediction accuracy of greater than 80% [13].
Homology modeling, 3D construction clarification and validation
We uploaded the created vaccine to the I-TASSER server (https://zhanggroup.org/I-TASSER) [24] in order to construct the structure prognosis. Then, to breed vaccine composition, we refine the vaccine from the Galaxyweb server (https://galaxy.seoklab.org/) [25]. The complete structure was downloaded from the portal, and the chosen structure was subsequently named based on the highest RMSD rate and effectiveness number. Using the PyMOL v2.3.4 software, we could observe the refined and refined formation practiced imaginatively. The ProSA-web accessory and Procheck demonstrated the significance of the Ramachandran plot and Z-point [26].
Molecular docking investigations
It highlights the key connections between protein model units and receptor units. For this docking study [27], we uploaded the completed vaccine model as ligand and the TLR4 protein as a receptor molecule to the ClusPro v2.0 site. The TLR4 receptor (PDB ID: 3W3M) was picked from the PDB website.
Molecular dynamics simulation study
The simulation of molecular dynamics was used to examine the physical motions of atoms and molecules and biophysical systems. This would allow us to assess the dynamics and safety of the vaccine-receptor fear [28]. The illusion had been removed from the iMODS website (http://imods.chaconlab.com).
Exempt protected rejoinder simulation
The whole construct was sent to the C-IMMSIM server (www.cbs.dtu.dk/services/C-ImmSim-10.1/) to see how the vaccine might affect the immune system [29]. As mentioned, we agreed that a 30-day break between application submissions would be the minimum acceptable gap [30].
Codon adaptation and in silico cloning technique
As the appearance of an alien gene in an organism is problematic, organism-specific codon optimization is required more frequently. Depending on the codon change, the JCat server released the construct. The modified course was evaluated based on the codon adaptation ratio (CAI) preference and guanine-cytosine content. In SnapGene v4.2, the body in silico cloning strategy was successfully implemented.
RESULTS
Best antigenic protein selection
Using UniProt server, different structural and non-structural protein sequences were retrieved to construct the vaccine. Based on antigenicity, the design protein scored with an antigenic point of 0.7055 (Vaxijen). Further analysis of these proteins’ amino acid sequences was done in order to determine CTL and HTL epitopes.
Possible HTL epitopes
A total of 195 epitopes, consisting of 15 amino acids, were identified using the IEDB server for mouse MHC-II alleles (IAb, IAd, IAs, IEb, IEd and IEs). We only selected among top five HTL epitopes based on antigenic scores to construct the final vaccine (Table 1).
Table 1. The selected HTL epitopes for the final vaccine construction.
Possible CTL epitopes
From NetCTL 1.2, server, structural, and non-structural protein CTL epitopes were predicted on the basis of antigenicity, allergenicity, toxicity and C. score. A number of 170 epitopes, where each consist of a length of nine amino acids CTL were predicted from the sorted out of spike protein. We took top five CTL epitopes based on antigenicity score (Table 2).
Table 2. The selected CTL epitopes for the final vaccine construction.
Possible LBL epitopes
All structural and non-structural proteins of linear B-cell epitopes were predicted from the iBCE-EL server, where they were shortlisted based on the prediction score. We chose only one out of a total of 36 B-cell epitopes that was shown to be antigenic, non-toxic, and non-allergenic (Table 3).
Table 3. The selected B cell epitopes for the final vaccine construction.
Vaccine construct and fundamental premises
The final vaccine was constructed with the selected 11 epitopes, which belong to three different classes, for example, 5 CTL, 5 HTL, and 1 LBL (Figure 2). All epitopes were joined by the AAY, GPGPG, and KK linkers. The TLR4 agonist 50S ribosome added in the beginning played the role of extra support to the immunogenicity to construct these 178 amino acid vaccines.
Physicochemical characteristics and immunological assessment
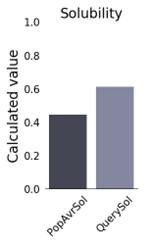
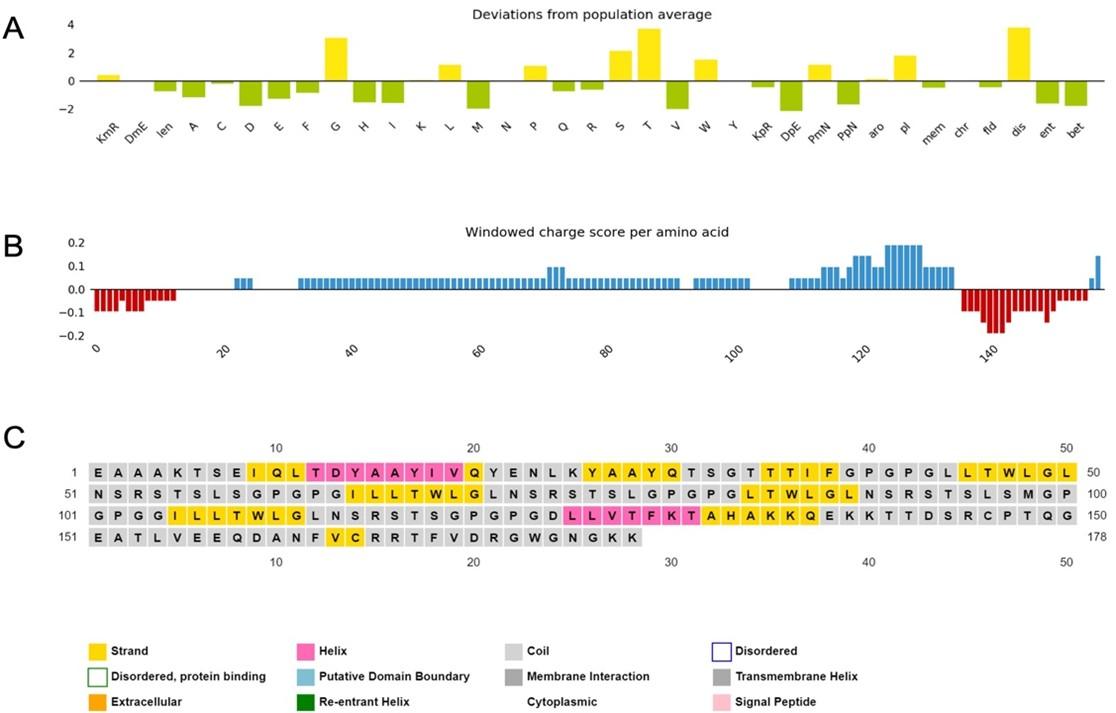
The physicochemical properties of the constructed vaccine were analyzed and documented, as shown in (Table 4). The constructed vaccine’s molecular weight was 18822.22 Da. In addition to the 178 amino acids, the theoretical isoelectric point (pI) was 9.36. Simultaneously, the vaccine had the formula C834H1319N231O259S3, an instability index of 35.33, an aliphatic index of 75.67, and a grand average of hydropathicity of -0.337. In addition, the constructed vaccine’s antigenicity was 0.7055, it was non-toxic, and its solubility was 0.614 out of 1 (Figure 3) indicating that it is highly soluble, and the windowed charge score and fold propensity score were depicted in Figure 4.
The a-helix, b-strand, and random coil secondary structures of the vaccine were evaluated using the SOPMA and PSIPRED servers (Table 5 and Figure 5A).
Table 4. Physicochemical characteristics of the construct.
Table 5. The secondary structural features of designated vaccine.
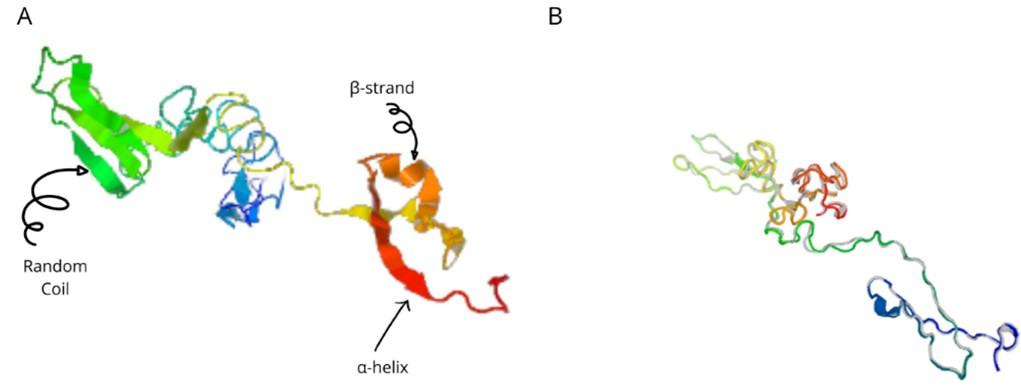
Tertiary structure, sophistication, and evaluation
The I-TASSER server (https://zhanggroup.org/I-TASSER) was used to obtain the best homology model (PDB id:01) among the top five modes. As advised by the server, we selected the lowest C-score (-3.38). Figure 5B depicts the designed vaccine’s tertiary structure. The vaccine demonstrated in the Ramachandran graph that it exceeds 93.8 percent in the significant region, with a GDT- score, RMSD, MolProbity 2.047, 1.276 Clash 1.3, and rotamers score of 0.0. This was determined by refining the model that we developed (Figure 6). The average Z score for the vaccine is -8.81 according to the Procheck online site, which was provided with Table 6, an additional file containing all findings, and the ProSA web server.
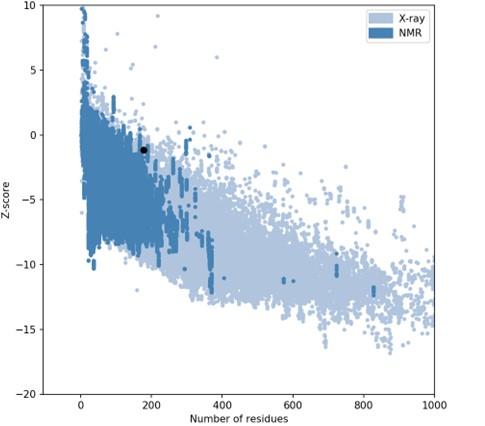
Table 6. The protein structure and overall structure geometry.
Molecular docking analysis
We evaluated the interaction between the refined model and immune receptor TLR-5 using a software-based simulation Clus Pro v2.0 site. It docked 30 models in various positions where the minimum energy value was determined using an additional file. Consequently, we placed model 2 in the dangling position with an energy score of -1401.2% (Figure 7).
Molecular dynamics simulation experiment
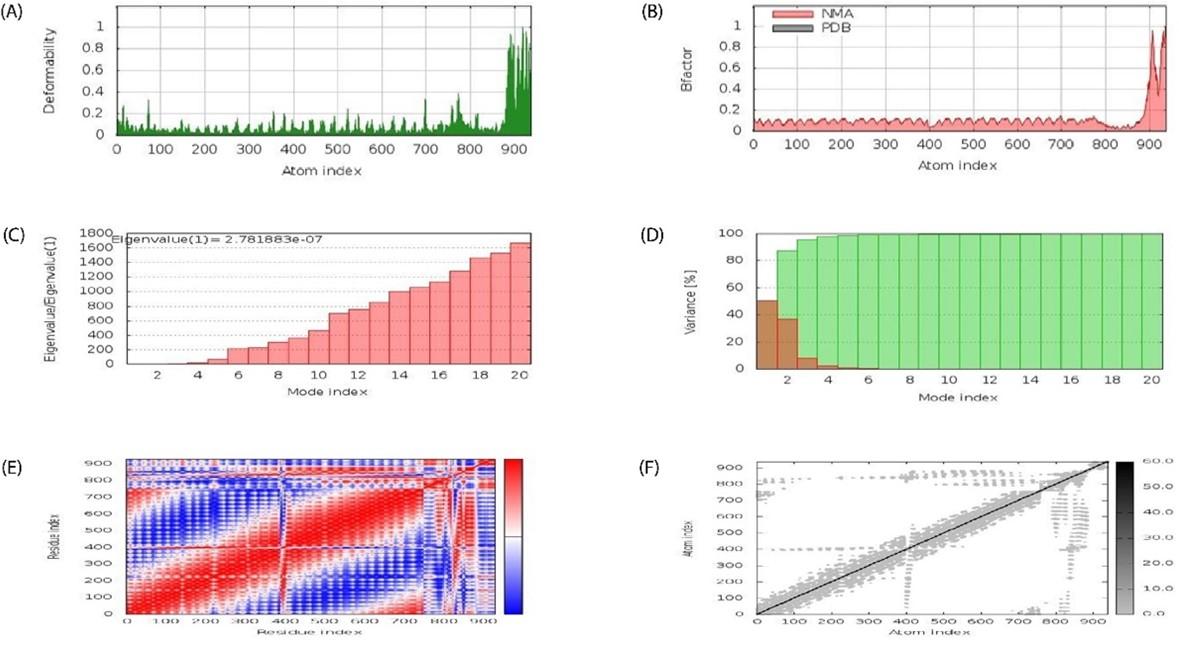
Docked complex was subjected to molecular dynamics using the iMODS server to examine stable interactions between ligand molecule and receptor (TLR-5) at the microscopic level. The average life B- factor map, Eigenvalues, Variance, Covariance map, and Elastic network were displayed on Figure 8A-F, respectively.
Exempt rejoinder simulation
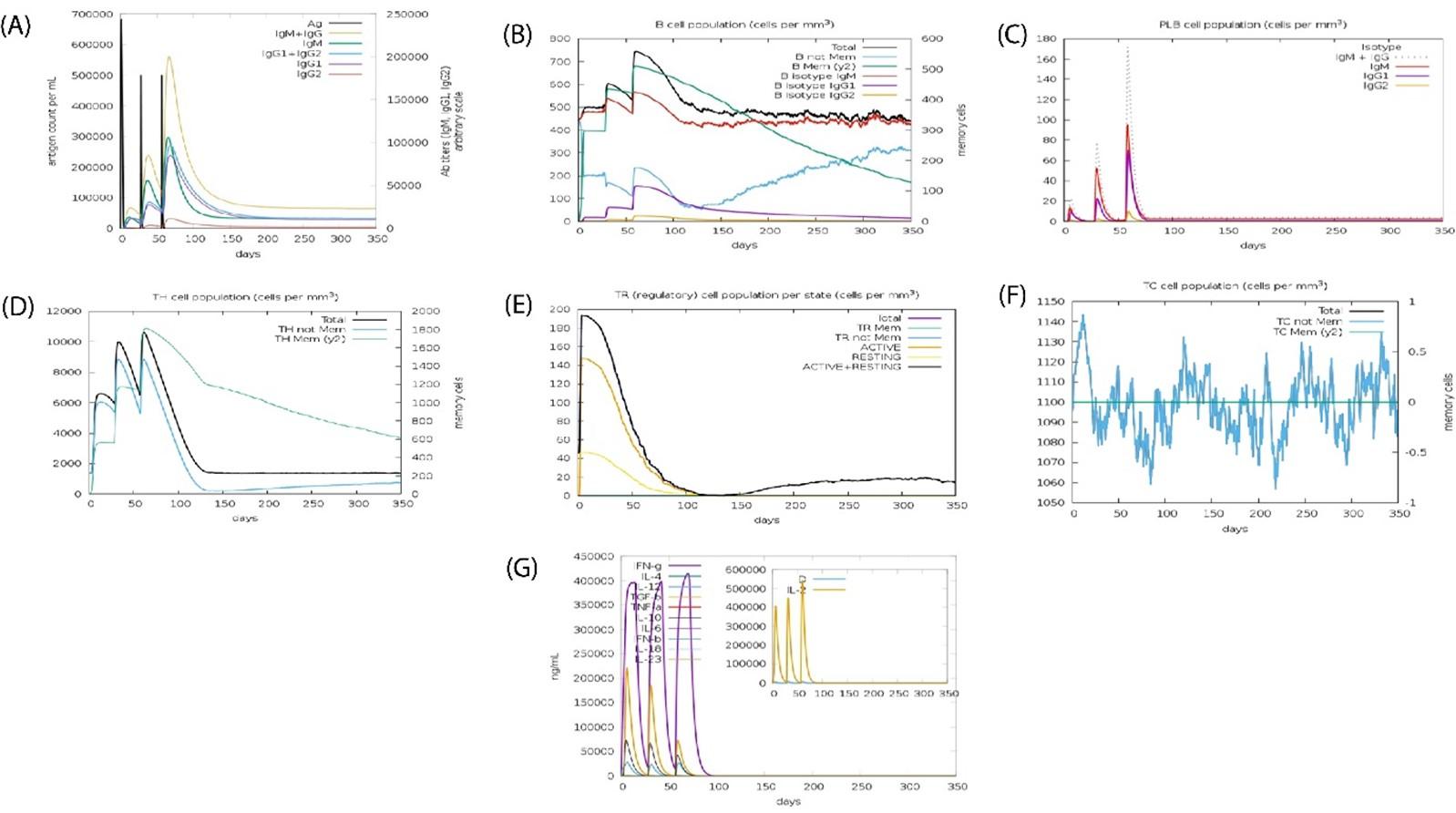
Special pathogens expressed produced actual immunological aspects confirmed (Figures 9A-G). Antigen and immunoglobulin values showed in Figure 9A, B lymphocytes cell; IgM and IgG in Figure 9B, plasma B lymphocytes count as sub-divided per isotope (IgM, IgG1, IgG2) in Figure 9C. The other value showed in Figure 9C to 9G.
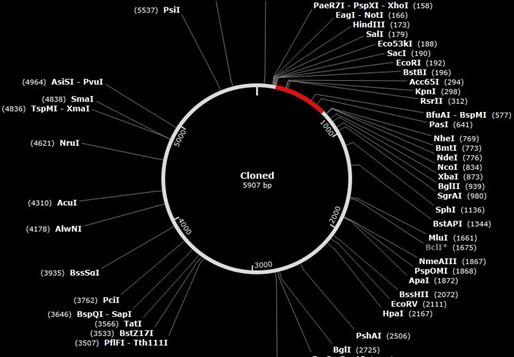
Codon evolution and in silico cloning
The codons in the created vaccine were optimized to develop their key factor according to the E. coli bacteria in JCat site. Lastly, the formed size of the vaccine cloning product is 5907 base pairs, and the vector was 5369, insert 546 base pairs (nucleotide base pair). Figure 10 depicts the cloned product that was manufactured.
DISCUSSION
An estimated 3.9 billion people worldwide are susceptible to dengue infection. In 129 nations, Asians shoulder the lion’s share of the burden or nearly 70 percent of the total [31].
In recent decades, millions of dengue cases have been reported annually, claiming the lives of a substantial section of the tropical and subtropical populations. In addition, the disease’s rapid global spread [13] makes dengue an ever-worsening global problem. dengue is a virus transmitted by mosquitoes, and different DENV serotypes can cause the disease. These serotypes (DENV-1-4) exhibit immunological cross-reactivity [31]. dengue treatment should stray as far as possible from “eliminating the pathogen and reducing confusion” [32]. Yet, no specific vaccination has been discovered due to the lack of an effective vaccine and the goal to limit vaccine adverse effects. A vaccine must be developed immediately to eradicate dengue from the human body. Various types of candidates are already available in the field of research, but none has yet been established. In this study, however, we introduce a multi-epitope-based vaccine based on computational methods for the Asian region. This novel strategy is required to combat this life-threatening public health condition. The S protein contributes to the viral host range, infectiousness, and human-to-human transmission. Therefore, we had to design the vaccine to target the S protein to reach the virus’s surface. We selected the CTL, HTL, and LBL epitopes in this study. We determined that the HTL was associated with the production of humoral and cellular immune responses and the HTL’s role in extending immunity and eliminating virus-infected cells.
Refining with the highest antigenic number, we selected one B-cell epitope from the top five models between HTL and CTL epitopes. The CTL, HBL, and LBL epitopes were attached to linkers to build a suitable vaccine. The constructed vaccine consists of 178 amino acids, and its physiological properties were established utilizing web resources. As the vaccine’s solubility was measured at 0.614 on a scale of 1, it was more soluble and would easily enter the E. coli host. Based on the vaccine’s physiological features, the theoretical PI value was determined to be 9.16 and fundamental. In addition, the physiochemical values of several metrics showed a higher possibility of effectiveness against the dengue virus. Web-based programs were used to analyze and forecast the homology modeling and three-dimensional structure of the manufactured vaccine. Using the PROCHECK server, the PDB file containing the final 3D structure of the vaccine was validated [31]. For the Ramachandran plot, we discovered a good overall predicted value of Z-score (-8.81) and the characteristics of the most preferred, accepted, and disallowed regions.
It is essential to establish the molecular basis of the TLR4 receptor’s effective immune response to develop the most efficient vaccination against MHC alleles (HLA-DRB1*04-01). Molecular docking between the peptide vaccine and the virus glycoprotein-binding favorable receptor of TLR4, which had the lowest energy score of -1401.2, suggested that the vaccination may inhibit infection. Furthermore, it revealed a potential close interaction between the modeled vaccine ligand and the TLR5 receptor surface [31]. Codon optimization was conducted to stabilize the built vaccine within the host for optimal multi-epitope vaccine generation [31], and codon optimization was performed. In silico cloning was performed using the JCAT web service [31] to optimize codons in a pET28a (+) vector to prevent codon bias. As long as we maintain the greatest degree of safety tools and procedures, we hope that the dengue virus will be eradicated if this vaccination instruction is followed in the laboratory.
CONCLUSIONS
Dengue is currently one of the most significant and life-threatening diseases in the world, and it is rapidly spreading. Bangladesh is one of the countries with the highest incidence of dengue transmission. Over the past few decades, dengue fever incidence has increased at an alarming rate. There is currently no effective and permanent treatment for dengue disease. Different approaches have been taken over the past few years, but none of them has yielded a definitive solution. In this study, a computational method was utilized to create a multi-epitope-based vaccine against the dengue virus.
Our designed vaccine possessed a high level of immunity and could bind with the immune receptor TLR4 to produce the greatest response against the dengue virus. As a result of this study, we hope that the designed vaccine may play a significant role in eradicating this rapidly spreading virus.
ACKNOWLEDGEMENT
The authors would like to thank the department of Biotechnology and Genetic Engineering, Bangabandhu Sheikh Mujibur Rahman Science and Technology University, Gopalganj, Bangladesh for supporting this research.
AUTHOR CONTRIBUTIONS
SRA, MIH and MSA designed the study. SRA, MIH and MSA performed the experiments, analyzed, and interpreted the data. SRA and MIH prepared the manuscript. MSA reviewed the manuscript. All authors approved the final version of the manuscript
CONFLICTS OF INTEREST
There is no conflict of interest among the authors.
References
- [1]Luo H, He J, Zheng K, Li L, za LJ-Z liu xing bing xue, 2002 undefined. Analysis on the epidemiologic features of Dengue fever in Guangdong province, 1990-2000. EuropepmcOrg n.d.
- [2]Soni A, Chugh K, Sachdev A, Gupta D. Management of dengue fever in ICU. Indian J Pediatr 2001;68.
- [3]Nhan NT, Phuong CXT, Kneen R, Wills B, Van My N, Phuong NTQ. Acute Management of Dengue Shock Syndrome: A Randomized Double-Blind Comparison of 4 Intravenous Fluid Regimens in the First Hour. Clin Infect Dis. 2001;32(2):204–13.
- [4]Damonte E, Matulewicz M, Cerezo A. Sulfated Seaweed Polysaccharides as Antiviral Agents. Curr Med Chem 2012;11.
- [5]Wagatsuma Y, Chowdhury M, Ahmed TU, Ashraf Uddin M, Nazmul Sohel S, Kittayapong P. Analysis of some Socio-demographic Factors Related to DF/DHF Outbreak in Dhaka City. 2000;24.
- [6]Pickett BE, Sadat EL, Zhang Y, Noronha JM, Squires RB, Hunt V, et al. ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res 2012;40:D593–8.
- [7]Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M, et al. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinformatics 2010;11.
- [8]Magnan CN, Zeller M, Kayala MA, Vigil A, Randall A, Felgner PL, et al. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 2010;26:2936–43.
- [9]Dimitrov I, Flower DR, Doytchinova I. AllerTOP – a server for in silico prediction of allergens. BMC Bioinformatics 2013;14.
- [10]Calis JJA, Maybeno M, Greenbaum JA, Weiskopf D, De Silva AD, Sette A, et al. Properties of MHC Class I Presented Peptides That Enhance Immunogenicity. PLoS Comput Biol 2013;9.
- [11]Larsen M V., Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics 2007;8:1–12.
- [12]Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GPS. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS One 2013;8.
- [13]Ali M, Pandey RK, Khatoon N, Narula A, Mishra A, Prajapati VK. Exploring dengue genome to construct a multi-epitope-based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci Rep 2017;7.
- [14]Nain Z, Abdulla F, Rahman MM, Karim MM, Khan MSA, Sayed S Bin, et al. Proteome-wide screening for designing a multi-epitope vaccine against emerging pathogen Elizabethkingia anophelis using immunoinformatic approaches. https://doi.org/101080/0739110220191692072 2019;38:4850–67.
- [15]Manavalan B, Govindaraj RG, Shin TH, Kim MO, Lee G. iBCE-EL: A New Ensemble Learning Framework for Improved Linear B-Cell Epitope Prediction. Front Immunol 2018;9:1695.
- [16]Dorosti H, Eslami M, Negahdaripour M, Ghoshoon MB, Gholami A, Heidari R, et al. Vaccinomics approach for developing multi-epitope peptide pneumococcal vaccine.
- [17]Olejnik J, Hume AJ, Mühlberger E. Toll-like receptor 4 in acute viral infection: Too much of a good thing. PLoS Pathog 2018;14:1–7.
- [18]Pandey, Rajan Kumar, Tarun Kumar Bhatt, and Vijay Kumar Prajapati. “Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein.” Scientific reports 8.1 (2018): 1-11.
- [19]Abdellrazeq GS, Fry LM, Elnaggar MM, Bannantine JP, Schneider DA, Chamberlin WM, et al. Simultaneous cognate epitope recognition by bovine CD4 and CD8 T cells is essential for primary expansion of antigen-specific cytotoxic T-cells following ex vivo stimulation with a candidate Mycobacterium avium subsp. paratuberculosis peptide vaccine. Vaccine 2020;38:2016–25.
- [20]Borthwick N, Silva-Arrieta S, Llano A, Takiguchi M, Brander C, Hanke T. Novel nested peptide epitopes recognized by CD4+ T cells induced by HIV-1 conserved-region vaccines. Vaccines 2020;8.
- [21]Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, et al. Protein Identification and Analysis Tools on the ExPASy Server. Proteomics Protoc Handb 2005:571–607.
- [22]Doytchinova IA, Flower DR. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 2007;8:1–7.
- [23]Buchan DWA, Minneci F, Nugent TCO, Bryson K, Jones DT. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res 2013;41:349–57.
- [24]Roy A, Kucukural A, Zhang Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat Protoc 2010;5.
- [25]Nugent T, Cozzetto D, Jones DT. Evaluation of predictions in the CASP10 model refinement category. Proteins Struct Funct Bioinforma 2014;82:98–111.
- [26]Wiederstein M, Sippl MJ. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 2007;35.
- [27]Kozakov, Dima, David R. Hall, Bing Xia, Kathryn A. Porter, Dzmitry Padhorny, Christine Yueh, Dmitri Beglov, and Sandor Vajda. “The ClusPro web server for protein–protein docking.” Nature protocols 12, no. 2 (2017): 255-278.
- [28]López-Blanco, José Ramón, José I. Aliaga, Enrique S. Quintana-Ortí, and Pablo Chacón. “iMODS: internal coordinates normal mode analysis server.” Nucleic acids research 42, no. W1 (2014): W271-W276.
- [29]Rapin N, Lund O, Bernaschi M, Castiglione F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS One 2010;5.
- [30]Castiglione, Filippo, Francesca Mantile, Piergiuseppe De Berardinis, and Antonella Prisco. “How the interval between prime and boost injection affects the immune response in a computational model of the immune system.” Computational and mathematical methods in medicine 2012 (2012):842329
- [31]Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, et al. The global distribution and burden of Dengue. Nature 2013;496.
- [32]Abdus Samad, Foysal Ahammad, Zulkar Nain, Rahat Alam, Raihan Rahman Imon, Mahadi Hasan & Md. Shahedur Rahman. Designing a multi-epitope vaccine against SARS-CoV-2: an immunoinformatics approach, Journal of Biomolecular Structure and Dynamics, 2022; 40:1, 14-30.