Validation and standardization of designed N gene primer-based RT-PCR protocol for detecting Peste des Petits Ruminants virus in goats
Abstract
The reverse transcriptase-polymerase chain reaction (RT-PCR) test is one of the most popular and specific diagnostic tests to easily recognize the Peste des Petits Ruminants virus (PPRV) genome in clinical samples. The sensitivity of the RT-PCR test depends on gene-targeted primer sets. The literature appears to be lacking in designing primers used in RT-PCR to detect PPRV genome in Bangladesh. This study aimed to develop an N gene based PPRV primer set, a standardized RT-PCR protocol, and its validity test by comparison with other available primers. A total of 70 clinical samples and 10 PPRV positive isolates were used in real-time RT-PCR and conventional RT-PCR using one pair designed primer set NF/NR and three pairs of published gene F1/F2, F1b/F2b and N1/N2. N gene based PPRV primer sets (NF/NR) were designed from a published sequence of PPRV (Accession number GQ122187.1). Statistical analysis was carried out. The designed N gene-based primer positive PPRV samples were sequenced and analyzed. The N gene-based primer sets were more sensitive to PPRV detection than F gene-based primer (P =.002) in the RT-PCR test. PPRV detects the highest (86%) of clinical samples in the RT-PCR test using a designed N gene-based primer. Sequence analysis showed that designed N gene-based the 402bp sequence of PPRV isolates is clustered with other Bangladeshi PPRV isolates and belongs to Lineage IV. New primers sets were designed from the conserved region of the N gene of PPRV. Designed primer sets successfully worked in real-time RT-PCR. The standardized RT-PCR protocol with the designed primer sets (NF/NR) can be used for the specific detection of PPRV from clinical samples.
INTRODUCTION
Peste des Petits Ruminants (PPR) is a highly contagious transboundary viral disease of sheep and goats that cause severe morbidity and mortality and has great economic concern [1-3]. PPR is caused by the PPR virus (PPRV), which is a negative sense single-stranded RNA enveloped virus belonging to the genus Morbillivirus of the family Paramyxoviridae [4]. The genome of PPRV, with a size of 15948bp, is organized into six transcriptional units. Each encodes at least one non-overlapping protein: the nucleocapsid (N), the matrix (M), the polymerase or large (L), the phosphoprotein (P), and two envelope proteins, hemagglutinin (H) and fusion (F). The P protein uses alternate expression strategies to code for two non-structural proteins viz., V and C [5]. Based on partial sequence analysis of the fusion (F) protein gene, PPRV isolates may be divided into four separate lineages [3]. Recent research showed that F and N gene polymerase chain reaction (PCR) methods were best suitable for detecting the PPR virus and assessing its lineages [6].
The F gene, which is responsible for viral fusion with the host cell, is thought to be the most critical element in virulence; hence the primers created for the F gene have been regarded as the most sensitive and popular method for diagnosing PPR [7]. The N gene is a major viral protein; the N gene-based primers were detected more in suspected samples than the F gene [8, 9]. Matrix (M) gene-targeted PCR had the highest sensitivity, followed by N, F, and H gene-based PCR [10]. FAO and OIE launched a PPR control and eradication program by 2030 around the world, like rinderpest eradication. Prompt and specific diagnosis of field outbreaks is critical for any disease control program to be effective, avoiding the potentially substantial economic harm from the outbreak. Early detection limits the spread of diseases and helps to control the diseases. Different molecular techniques such as RT-PCR (reverse transcription-PCR), real time PCR (RT-qPCR), multiplex PCR, and loop-mediated isothermal amplification (LAMP) have been established to easily recognize the PPRV genome in clinical samples [10]. Traditional RT-PCR tests use gel electrophoresis to identify PCR products, which adds time and work to the process. Real-time PCR (RT-qPCR) tests provide various benefits over conventional RT-PCR assays, such as minimizing contamination by completing both amplification and analysis in a closed system, providing quantitative RNA measurements, and being faster and more sensitive to execute.
Since 1995, numerous RT-PCR assays targeting F, M, N, H etc., proteins have been developed for the quick and specific detection of PPRV [8, 11]. For the confirmatory diagnosis of PPRV, most lab use RT-PCR protocol targeting F and N gene-based PCR [8]. The N gene codes for an internal structural protein, and N gene mRNAs are the virus’s most abundant transcripts, making it an appealing target for the creation of a highly sensitive diagnostic test. Indeed, since their polymerases lack a proof-reading function, RNA viruses are known to have a high incidence of nucleotide substitution errors [12]. To achieve successful amplification and detection of such viruses, primers need to be constructed by finding conserved regions in published sequences of viral isolates from various geographical areas; this technique reduces the danger of false-negative results caused by primer-template mismatch [9]. The sensitivity of the RT-PCR test also depends on gene-targeted primers sets [13]. The available published primers were designed more than ten years ago [8, 9]. However, the literature lacks a designed N gene primer-based RT-PCR for early and specific PPRV detection in Bangladesh. From this aspect of view, the study aims to design a new N gene-targeted primers set and validate its by conventional and real-time PCR; and compare it to other currently available genes for the detection of PPRV in field samples in Bangladesh.
MATERIALS AND METHODS
Ethical approval
The committee of ethical standards, Bangladesh Agricultural University Research System (BAURES), has been approved the research work with reference number BAURES/2020ESRC/VET/08, dated: 03.06.2021.
Study plan
A schematic diagram of the study is represented in Figure 1.
Collection of clinical samples
A total of seventy (70) samples (thirty samples from goats at the different slaughterhouse and forty nasal swab samples from clinical PPR suspected goats) were aseptically collected from the Mymensingh division during the winter season (November 2020 to February 2021). Tiny pieces of grossly visible pneumonic lungs and fibrinous covered spleen from each slaughtered goat were collected in a cryotube, snap-frozen, and immediately shifted to a -20ºC freezer for further study. Nasal swabs from clinically PPR suspected goats were collected in 1X PBS (Phosphate buffer saline) containing tube and immediately shifted to the normal refrigerator at 4ºC. RNA was extracted as early as possible.
RNA extraction
Approximately 100mg of lung tissue and 100 mg of spleen from goats were individually triturated using an automatic tissue lyser (Qiagen tissue lyser-II, USA). At first, tissues were taken into a sterile eppendorf tube containing 500µl (1X) PBS, 5% Penstrep with the metal bidder, and grinding for 5 minutes. Then the tissue homogenates were centrifuged at 5000 rpm for 5 minutes. After that, the supernatant fluid was collected in a separate eppendorf tube. According to manufacturer instructions, RNA was extracted from the tissue fluid and swab fluid using a commercially available RNA extraction kit (Viral RNA/DNA pure link mini kit, Invitrogen, USA).
The purity and concentration of extracted RNA were measured at 260nm/ 280nm in a NanodropTM spectrophotometer (IAEA, Scibersdoff, Vienna). A 260nm/ 280nm ratio of ~2.0 for RNA was considered “pure” and used for RT-PCR assays to detect the specific gene of the PPRV. The sample viral RNA ranged from 20ng – 50ng/µl was used for real-time PCR and 50ng – 100ng/µl viral RNA was used for conventional RT-PCR.
Oligonucleotide primers
Two pairs of F gene-based published primer F1/F2, F1b/F2b and a pair of N gene-based published primer N1/N2, were used in this study. In addition, a primer pair was designed targeting N gene-based NF/NR primers from a published PPRV sequence (GenBank Ac. No. GQ122187.1). These four pairs of primer sets were obtained from Macrogen, Korea (Table 1). The designed NF/NR primer sets sensitivity and specificity were compared with the other three pairs of published primer sets.
Table 1. Oligonucleotide primers set used for the PPRV detection.
Standardization of RT-PCR protocol with the designed NF/NR primer
Locally ten (10) PPR virus-positive isolates in our lab were used as the positive control. Grossly healthy lung tissue RNA with the required primers and the master mix was used as the negative control. Four pairs of primer set such as F1/F2, F1b/F2b, N1/N2 and designed NF/NR genes were used in conventional RT-PCR to detect PPRV from positive isolates.
Detection of PPRV from clinical samples by using designed NF/NR primer on real-time RT-PCR
Designed N gene primer sets (NF/NR) were used in real-time PCR. Early detection of the PPR virus in the sample was performed by real-time PCR. RT-qPCR was done on an AB 7500 Fast Real-time PCR Machine using Luna® Universal One-step RT-qPCR (New England Biolab Ltd, Inc., USA). The master mix was prepared as a reaction volume of 20µl comprising Luna Universal One-step reaction mix (2X) 10 µl, Luna Warm Start® RT enzyme mix (20X) 1 µl, forward primer NF (10pmol/µl) 1 µl, reverse primer NR (10pmol/µl) 1 µl, nuclease-free water 5µl and the template RNA 2 µl. Forty cycles of RT-qPCR amplification were carried out for NF/NR gene using reverse transcription 55ºC for 10 min, initial denaturation 95ºC for 1 min, denaturation 95ºC for 10 sec, annealing temperature 55ºC for 30 sec, extension 60ºC for 30 sec, melting curve 95ºC for 15 sec, 60ºC for 1 min and 95ºC for 30 sec. RNA from apparently healthy lung tissues was used as the negative control. The results were presented as the Ct value for each sample, determined from the amplification plot.
Detection of PPRV from clinical samples by using designed NF/NR primer on conventional RT-PCR
Comparisons among four pairs of the gene were carried out by conventional RT-PCR. For cost effective analysis, conventional RT-PCR uses real-time PCR positive field samples in comparative gene analysis. The RT-PCR reaction was performed in an oil-free thermal cycler (Proplex gradient PCR, USA). The RT-PCR was carried out with a 50µl reaction volume using one Taq one-step RT-PCR kit (New England, Biolab Ltd). The master mix comprises one Taq one-step reaction mix(2X) 25 µl, one Taq one-step enzyme mix (25X) 2µl, forward primer (10pmol/µl) 2µl, reverse primer (10pmol/µl) 2µl, nuclease-free water 17µl and template RNA 2µl.
The RT-PCR amplification of the selected gene of the PPR virus was started with the reverse transcription at 48°C for 20 min. Then the initial denaturation was carried out at 94°C for 1 min followed by 40cycles for the NF/NR gene and 35 cycles for N1/N2, F1/F2 and F1b/F2b genes. The amplification reaction consists of denaturation at 94°C for 15sec, annealing temperatures at 55°C for 30sec (NF/NR and N1/N2 genes), 60°C for 30sec (F1/F2 and F1b/F2b genes), and elongation at 68°C for 1min and final elongation at 68°C for 5min. RNA from healthy lung tissues with required primers and master mix was used as the negative control. The RT-PCR products were electrophoresed (WSE-1710Submerge-Mini2322100, China) in 1.5% agarose gel containing ethidium bromide (0.5μg/ml), and images were captured in a transilluminator (Alpha imager, USA). A 100bp DNA ladder (TackIT, Invitrogen, USA) was used in the agarose gels to evaluate the size of amplicons.
Sequence analysis of designed NF/NR gene primer and NF/NR gene specific field samples
The designed NF/NR gene based PPRV positive PCR products (03 field samples) were sequenced from a commercial source (Macrogen Inc, Korea). This sequence data was first identified by the online Basic Local Alignment Search Tool (BLAST) and downloaded the PPR virus gene isolated from different countries in different years retrieved from the National Center for Biotechnology Information (NCBI) gene bank. Theses sequences were aligned using BioEdit 7.0.5 [14]. CLC Sequence Viewer 8 software helped to perform the multiple alignments of the sequences using UPGMA, Kimura 80, and Maximum Likelihood (MJ) tests to perform phylogenetic analysis [15]. The phylogenetic tree reliability was checked by 100 replicates of Bootstrap values [16].
The newly designed forward (NF) and reverse primers (NR) and other twelve complete N gene of PPRV isolates (China, India, Morocco, and Egypt) downloaded from NCBI were aligned using CLC sequence viewer 8.0.
Statistical analysis
The Pearson chi-square test for independence relatedness of the variants and descriptive analysis of the four pairs primer test data was carried out by using SPSS 22 (IBM corporation, United States) to estimate the significant differences among different genes used to detect PPRV. A value of P ≤ 0.05 was considered significant at a 95% confidence interval.
RESULTS
Standardization of RT-PCR protocol with the designed NF/NR primer
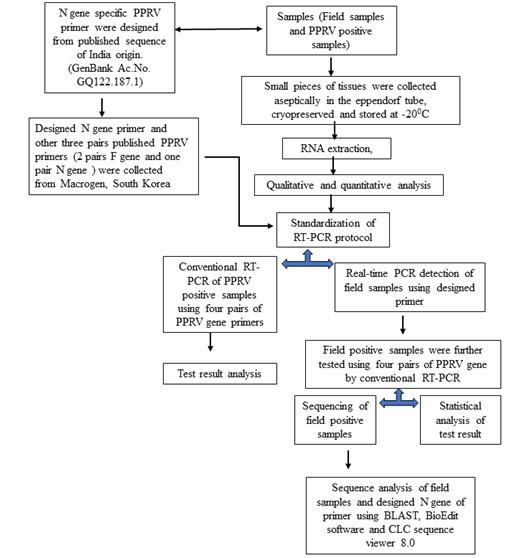
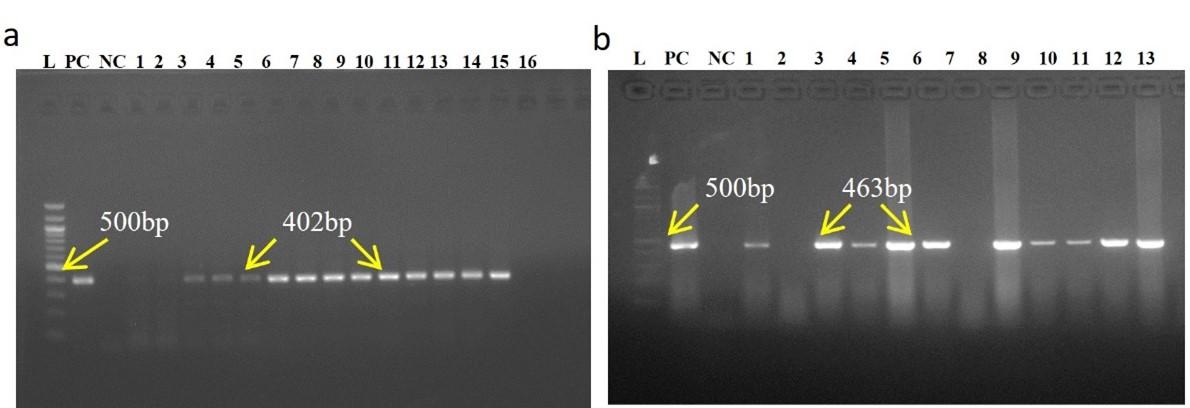
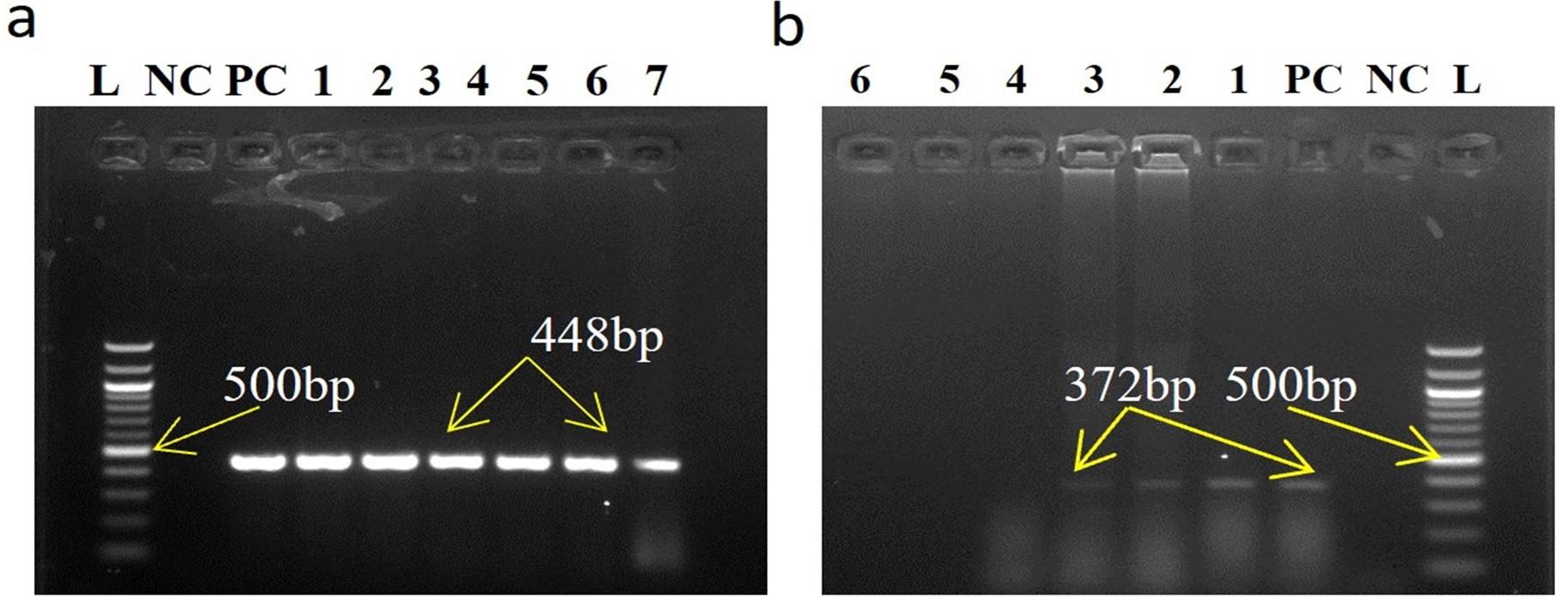
Standardized RT-PCR protocol and comparative analysis of four pair of primer sets sensitivity to PPRV positive isolates were done in conventional RT-PCR. All PPRV positive isolates (10) were successfully amplified by a newly designed NF/NR primer and produced 402bp amplicons (Figure 2). Out of ten (10) PPRV isolates, eight (08) isolates were showed a positive response to N1/N2 primer produced 463bp amplicons, six (06) isolates positive in F1b/F2b primer produced 448bp amplicons, and only four (04) isolates showed positive in F1/F2 gene produced 372bp amplicons. Negative samples RNA did not generate any amplicon by conventional RT-PCR.
Detection of PPRV from clinical samples by using designed NF/NR primer on real-time RT-PCR
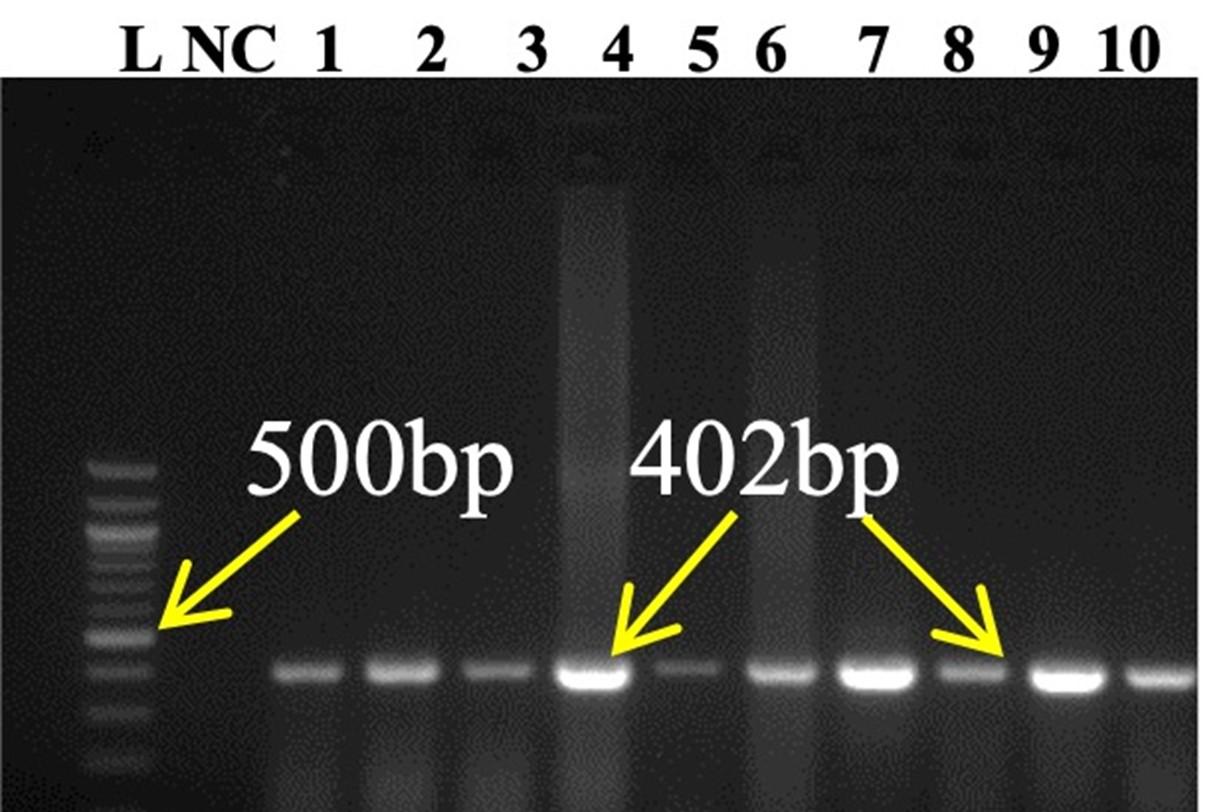
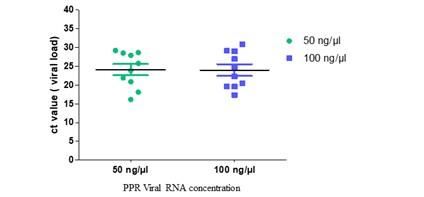
Out of seventy (70) field samples, thirty (30) samples showed positive amplification during real-time PCR. PPRV positive samples showed Ct value ranging from 16-25 (Figure 3). The 50ng/µl of PPRV RNA Ct mean value was 24.10 and 100ng/µl of PPRV RNA Ct mean value was 23.98. The PPR viral RNA 50ng/µl was successfully amplified in the RT-qPCR test.
Detection of PPRV from clinical samples by using designed NF/NR primer on conventional RT-PCR
Out of 30 RT-qPCR based PPRV positive clinical samples analyzed, newly designed N gene (NF/NR) based RT-PCR detected the highest number in 26 samples. NF/NR gene-based RT-PCR produced amplicon as 402bp (Figure 4, arrow, a). N1/N2 based RT-PCR detected in 25 samples. N1/N2 gene-based RT-PCR produced amplicon as 463bp (Figure 4a, arrow). F1b/F2b based RT-PCR detected in 23 samples. F1b/F2b gene-based RT-PCR produced amplicon as 448bp (Figure 5a, arrow). F1/F2 based RT-PCR detected in 13 samples. F1/F2 gene-based RT-PCR produced amplicon as 372bp (Figure 5b, arrow).
Sequence analysis of designed NF/NR gene specific clinical samples and designed NF/NR primer
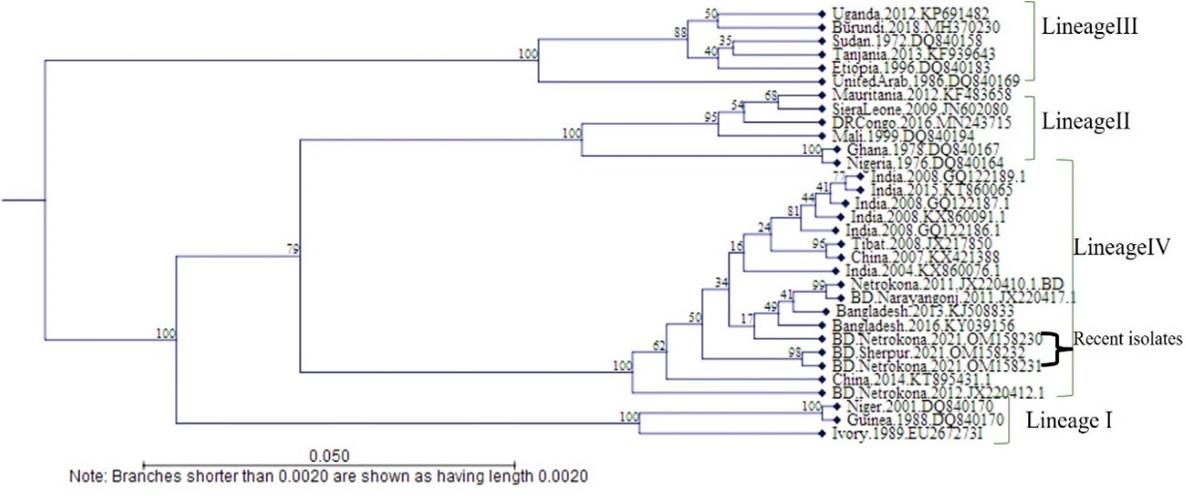
The designed NF/NR gene-targeted three (3) PPRV field samples were first identified by BLAST search and showed a percent identity 97-99% with other Bangladeshi, Indian and Chinese isolates. These three PPRV isolates were then submitted to GenBank and got the accession number OM158230, OM158231, and OM158232. The phylogenetic tree was made based on the designed N gene targeted 402bp sequence of PPRV isolates clustered on other downloaded isolates of Bangladesh along with India, China, and other Asian isolates belonging to Lineage 4 (Figure 6). The designed NF/NR gene (Designed from GenBank Ac no. GQ122187.1) were aligned with other downloaded twelve complete N gene sequences of China, India, Morocco, and Egypt, showed 100% homology (Figure 7). Sequence analysis revealed that the NF/NR genes were designed from the conserved region of the N gene of PPRV.
Comparative analysis of primers
Among four pairs of N and F gene-based RT-PCR test result, the N gene primers-based RT-PCR were more sensitive to PPRV detection from clinical samples followed by F genes (P =.002) (Table 2).
Table 2. Comparative analysis of four pairs of primers by conventional RT-PCR.
DISCUSSION
PPR disease is a devastating illness in sheep and goats in Africa, the Middle East, and Asia. PPR yields a detrimental effect on food security and the livelihoods of impoverished farmers who are the primary producers of sheep and goats. Like rinderpest eradication from the world, FAO and OIE launched a program called PPR global control and eradication strategy by 2030. The effectiveness of any disease control program depends on the use of reliable and strong diagnostic technologies. Many diagnostic procedures have been performed like clinical diagnosis, ELISA (Enzyme linked immunosorbent assay), virus isolation, VNT (Virus neutralization test), immunocapture, AGID (Agar gel immunodiffusion test), LAMP (Loop mediated isothermal amplification), real-time PCR, RT-PCR [8, 10, 17]. Nucleic acid-based diagnostic tests are more sensitive and specific [7]. PPRV nucleic acid may be detected in clinical samples using RT-PCR. It has been used to confirm and differential diagnosis of PPRV by focusing on the F and N genes of the virus [8]. RNA viruses are notorious for committing nucleotide substitution mistakes due to the lack of a proof-reading function in their polymerases [12].
Finding conserved areas in published viral sequences from different locations of the world is essential for the successful amplification and detection of such viruses. This strategy lowers the risk of false-negative findings caused by primer-template mismatch [9]. In this study, newly developed NF/NR gene primers were designed from the published N gene of the PPRV sequence (GenBank Ac. No. GQ122187.1), an isolated PPRV from India that is belongs to lineage IV. To assess the genetic similarity and divergence of designed NF/NR primers with twelve other complete N gene of published PPRV isolates from China, India, Morocco and Egypt revealed 100% homology. The circulating PPRV in Bangladesh belongs to lineage IV. In this study, the test positivity of designed NF/NR primers is the highest (86%) in clinical sample detection. The designed NF/NR genes showed a 100% positive response in the case of previously positive PPRV isolates
Several conventional RT-PCR systems have been described for the detection and identification of PPRV. On the other hand, real-time PCR offers numerous benefits over conventional RT-PCR, including being faster and more sensitive and being done in a single tube, preventing potential cross-contamination during sample preparation for gel electrophoresis [18-20].
Quantitative analysis and identification of PPRV have been accomplished using real-time PCR technology. RT-qPCR, which was created recently, has become the most well-known technique for detecting the Paramyxoviridae family. This advanced technique of RT-qPCR dominates the traditional techniques [21]. One-step real time quantitative reverse transcription PCR (RT-qPCR) based upon the TaqMan probe to detect PPRV was developed and stated that RT-qPCR enhanced the PPRV detection rate from 46.7% to 73.33%, compared to the conventional RT-PCR method [19]. In this study, one step real-time RT-qPCR was used to detect PPRV in suspected samples. A designed NF/NR gene-based primer successfully worked in real-time RT-qPCR with a greater rate. Real-time PCR provides sensitive and specific detection of all PPRV lineages, including those circulating in Africa, the Middle East, and Asia [22, 23]. Real-time PCR could identify 20% more positive findings with low viral RNA levels than conventional RT-PCR [22]. In this study, only 50-100ng/µl RNA samples were used for PPRV detection, whereas 100-500ng/ µl RNA samples were used in conventional RT-PCR. Low viral RNA was amplified in real-time PCR.
The F gene responsible for the fusion of the virus with host cell is the important factor responsible for virulence. RT-PCR based on the F gene has acquired a lot of popularity [7]. However, PCR based on other gene targets has recently been accessible [24]. The PPR-specific primers F1 and F2 produced by the researchers mentioned above amplify a 372 bp region of the PPRV F gene between positions 777 and 1148 nucleotides [9]. In this study, out of 30 PPRV positive field sample, F1/F2 gene of PPRV was detected in 14 samples (46%) and 16 samples (53%) failed to generate a positive response. However, the F1/F2 primer set did not function well on some samples; therefore, the researchers employed a pair of another F gene-based primers (F1b/F2b) situated just outside the F1/F2 area to amplify the region between 760 and 1207 nucleotides of the F gene, resulting in a 448bp amplicon [3]. This study revealed that out of 30 samples tested, the F1b/F2b gene of PPRV was detected in 21 samples (70%) whereas only 7 samples (23%) failed to generate a positive response. The F1b/F2b gene-based primer in RT-PCR is more sensitive to PPRV detection than F1/F2 gene primer in RT-PCR [3]. N gene-specific RT-PCR is more sensitive than F gene-based RT-PCR [25]. Recently, many promising attempts have been made to target the N gene for PCR-based PPRV detection [8, 26]. The genetic characterization of Indian PPRV was done based on a new set of primers (N1/N2) that targeted the N gene at 1208-1226 and 1670-1652 base position, respectively, and made an amplicon of 463 bp. The researchers found that the newly designed N gene primers of PPRV are more sensitive than the F gene-based primers (F1/F2) in RT-PCR [9]. Though they have been proven to be more sensitive, the N gene-based primers were not yet gained widespread recognition in the diagnosis of PPRV. Primers N1/N2 were created in-house using the GenBank sequences.
In this study, considering the lower sensitivity of the F gene based PPRV test, N gene-based primer NF/NR gene were designed to detect PPRV genome in clinical samples. This study found that N gene-based primers more sensitive than F gene-based primers set in the RT-PCR test. Nucleoprotein (N) gene-based primers NF/NR (GenBank Ac.no. GQ122187.1, India, 2008) targeting the N gene position of PPRV at 1130-1150, 1532-1509 base positions, respectively generate 402bp amplicons for specific and sensitive detection of PPRV from clinical samples. These N gene-based primers were designed from the published sequence of PPRV from a neighboring country belongs to Lineage IV. The circulating PPRV in Bangladesh is belongs to Lineage IV [27, 28]. This study revealed that out of 30 positive samples tested, NF/NR gene primer produced a positive response highest in 26 samples (86%). The rest 4 positive samples gave negative response in conventional PCR due to low concentration of viral RNA. On the other hand, N1/N2 gene primers have positive responses in 25 positive cases (83%). The sensitivity of NF/NR and N1/N2 gene was much better than that of the F gene [8, 9]. The N gene codes for an internal structural protein and N gene mRNAs are the virus’s most abundant transcripts, making it an appealing target for creating a susceptible diagnostic test.
PPRV isolates are genetically grouped into four separate lineages (I, II, III and IV) [3, 24]. Lineages I and II are only found in West Africa, but lineage III has been identified in Ethiopia, Arabia (Oman, Yemen), and southern India (Tamil Nadu). Lineage IV of PPRV was circulating in the Middle East, Arabia, and the Indian subcontinent. The literature showed that F and N gene-based RT-PCR methods were best suitable for detecting PPR virus and assessing its lineages [6]. The recent South Asian PPR viruses from India and Bangladesh are closely related. However, there was very minimal sequence divergence in this lineage, up to a maximum of 3.5% at the nucleotide level [24]. In this study phylogenetic analysis revealed that designed N gene specific PPRV clinical samples showed 97-99% similarity with other Bangladeshi isolates and other Asian isolates like India and China; and this PPR virus belongs to lineage IV.
CONCLUSIONS
A newly developed RT-PCR protocol can be used for the specific detection of PPRV by using primers NF/NR designed from a published sequence (GenBank Ac.no. GQ122187.1). NF/NR primers successfully amplified PPR virus in PPRV positive isolates and suspected clinical samples in real-time PCR and conventional RT-PCR. Sequence analysis of the cDNAs of NF/NR gene primers revealed that these primer sets are designed from the conserved region of the N gene of PPRV. The phylogenetic tree based on the 402bp sequence of designed N gene of recent PPRV isolates clustered on other downloaded isolates of Bangladesh and India, China, and other Asian isolates belongs to Lineage IV. Comparison to other published gene revealed that N gene-based RT-PCR is more sensitive to PPRV than F gene-based RT-PCR. F1/F2 gene-based RT-PCR may provide false-negative findings. PPRV can be identified using a one-step real-time PCR method, which is the quickest, sensitive, and most specific method. Very low amount of RNA can be used to detect PPRV by real-time PCR than conventional RT-PCR.
ACKNOWLEDGEMENT
Thanks to the National Agricultural Technology Program (NATP) Phase II, Bangladesh Agricultural Research Council (BARC) through Bangladesh Agricultural University Research System (BAURES) for funding the research (Grant ID 139/2018-2021). Special thanks to Dr. Jayedul Hassan, Associate Professor, Department of Microbiology & Hygiene for helping in the sequence analysis.
AUTHORS CONTRIBUTION
SS, MP and MAHNAK: Designed the study, generated research fund, and tuned the manuscript for submission. SS, NS and MRI helped in collecting samples and did Real time PCR, RT-PCR and gel electrophoresis. SS did the real-time PCR. SS and NS did the statistical analysis. SS drafted the manuscript. SS, MP, NS and MAHNAK revised the final manuscript. All authors read and approved the final submission.
CONFLICTS OF INTEREST
There is no conflict of interest among the authors.
References
- [1]Khan HA, Siddique M, Arshad MJ, Khan QM, Rehman SU. Seroprevalence of peste des petits ruminants (PPR) virus in sheep and goats in Punjab province of Pakistan. Pakistan Vet. J. 2007; 27(3): 109-112.
- [2]Asim M, Rashid A, Chaudhary AH, Noor MS. Production of homologous live attenuated cell culture vaccine for the control of Paste des petits ruminants in small ruminants. Pakistan Vet. J. 2009; 29(2): 72-74.
- [3]Dhar P, Sreenivasa BP, Barrett T, Corteyn M, Singh RP, Bandyopadhyay SK. Recent epidemiology of peste des petits ruminant’s virus (PPRV). Vet. Microbiol. 2002; 88(2): 153–159.
- [4]Gibbs EP, Taylor WP, Lawman M, Bryant J. Classification of the peste-des petits-ruminants virus as the fourth member of the genus Morbillivirus. Intervirology. 1979; 11: 268-274
- [5]Muthuchelvan D, Sanyal A, Sarkar J, Sreenivasa BP, Bandyopadhyay SK. Comparative nucleotide sequence analysis of the phosphoprotein gene of peste des petits ruminant’s vaccine virus of Indian origin. Res. Vet. Sci. 2006; 81(1): 158–164.
- [6]Pandey AK, Muglikar DM, Mhase PP, Pawade MM, Daphal SN, Pawar PD. F Gene and N Gene based Reverse Transcription PCR for Molecular Characterization of Peste des Petits Ruminants Virus. Indian J Anim Res. 2020; B3727(1-5) http://dx.doi.org/ 10.18805/ijar.B-3727.
- [7]Forsyth M, Orsyth TM, Barret T. Detection and differentiation of rinderpest and peste des petits ruminants’ viruses in diagnostic and experimental samples by polymerase chain reaction using P and F gene-specific primers. Virus Res. 1995; 39: 151-163.
- [8]Couacy-Hymann E, Roger F, Hurard C, Guillou J, Libeau G, Diallo A. Rapid and sensitive detection of peste des petits ruminant’s virus by a polymerase chain reaction assay. J. Virological methods. 2002; 100: 17-25.
- [9]Kerur N, Jhala MK, Joshi CG. Genetic characterization of Indian peste des tion of Indian peste des petits ruminants virus (PPRV) by sequencing and phylogenetic an etits ruminants virus (PPRV) by sequencing and phylogenetic analysis of fusion protein and lysis of fusion protein and nucleoprotein gene segments. Res. Vet. Sci. 2008; 85: 176-183.
- [10]George A, Dhar P, Sreenivasa BP, Singh RP, Bandyopadhyay SK. The M and N genes-based simplex and multiplex PCRs are better than the F or H gene-based simplex PCR for Peste-des-petits-ruminants virus. Acta Virol. 2006; 50(4): 217–22.
- [11]Balamurugan V, Sen A, Saravanan P, Singh RP, Singh RK, Rasool TJ, Bandyopadhyay SK. One-step multiplex RT-PCR assay for the detection of peste des petits ruminants virus in clinical samples. Vet. Res. Commun. 2006; 30: 655–666.
- [12]Steinhauer DA, Holland JJ. 1986. Rapid evolution of RNA viruses. Annu. Rev. Microbiol. 41, 409–433.
- [13]Bhuiyan AR, Rahman MM, Begum JA, Islam MR, Chowdhury EH. Comparison of genes as target for molecular diagnosis of peste des petits ruminants in goats. Bangladesh Vet. 2012; 29(2) : 56 – 62
- [14]Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Nucleic Acids Symposium Series. 1999; 41(41): 95-98. [London]: Information Retrieval Ltd., c1979-c2000.
- [15]Nei M, Kumar S. Molecular Evolution and Phylogenetics. New York: Oxford University Press. 2000; 123–333.
- [16]Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013; 30:2725–2729.
- [17]Tiwari A. Prevalence of Peste des petits ruminants (PPR) virus in small ruminants of Gujarat and its characterization by RT- PCR/ RFLP and SSCP profiles. M.V.Sc. thesis submitted to Anand Agricultural University, Anand, India. 2004.
- [18]Aguero M, Sanchez A, San Miguel E, Gomez-Tejedor C, Jimenez-Clavero MA. A real-time TaqMan RT-PCR method for neuraminidase type 1 (N1) gene detection of H5N1 Eurasian strains of avian influenza virus. Avian Dis. 2007; 51: 378–381.
- [19]Bao J, Li L, Wang Z, Barrett T, Suo L, Zhao W, Li J. Development of onestep real-time RT-PCR assay for detection and quantitation of paste des petits ruminants virus. J. Virol. Method. 2008; 148 (1): 232-36.
- [20]Shaw AE, Reid SM, Ebert K, Hutchings GH, Ferris NP, King DP. Implementation of a one-step real-time RT-PCR protocol for diagnosis of foot-and-mouth disease. J. Virol. Methods. 2007; 143: 81–85.
- [21]Grant JR, Banyard AC, Barrett T. Real-time RT-PCR assays for rapid and differential detection of dolphin and porpoise morbillivirus. J. Virol. Method. 2009; 156(1-2): 117- 23.
- [22]Kwiatek O, Ali YH, Saeed IK, Khalafalla AI, Mohamed OI, Obeida A, Abdelrahman MB, Osman HM, Taha KM, Abbas Z, El Harrak M, Lhor Y, Diallo A, Lancelot R, Albina E, Libeau G. Asian lineage of peste des petits ruminants virus, Africa. Emerg. Infect. Dis. 2010; 17: 1223–1231.
- [23]Batten C, Banyard AC, King DP, Henstock MR, Edwards L, Sanders A, Buczkowski H, Oura CAL, Barrett T. A real-time PCR assay for the specific detection of peste des petits ruminants virus. J. Virol. Methods. 2011; 171 (2): 401–404.
- [24]Shaila MS, Shamaki D, Forsyth MA, Diallo A, Goatley L, Kitching RP, Barrett T. Geographaic distribution and epidemiology of peste des petits ruminants virus. Virus Res. 1996; 43: 149-153.
- [25]Mahajan S, Agrawal R, Kumar M, Mohan A, Pande N. Comparative evaluation of different F and N gene based reverse transcription polymerase chain reaction for molecular detection of peste des petits ruminants virus from clinical samples. Vet. Arhiv. 2014; 84: 485-492.
- [26]George AA. Comparative evaluation of different gene targets for PCR diagnosis of PPR. M.V.Sc. Thesis, Deemed University IVRI, Izatnagar, India. 2002.
- [27]Rahman MM, Parvin R, Bhuiyan A, Giasuddin M, Chowdhury SM, Islam MR, Chowdhury EH. Genetic characterization of Peste des petits ruminants virus circulating in Bangladesh. British J Virol. 2016; 3(4): 115–122.
- [28]Mohammed ZR, Najmul H, Emily G, Sadia A, Mozaffar G O et al. Epidemiology and genetic characterization of Peste des petit ruminants virus in Bangladesh. Vet. Med. Sci. 2018; 4(3):161–171.