Prediction of B-cell and T-cell epitopes in the spike glycoprotein of SARS-CoV-2 in Bangladesh: An in-silico approach
Abstract
To combat highly infectious Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), scientists and researchers are toiling hard globally to develop effective drugs and vaccines. By exploring the structural proteins of SARS-CoV-2 can be a feasible way to find an effective vaccine. In this study by using in-silico tools, we recommended B-cell and T-cell epitopes of spike protein from a Bangladeshi isolate which can be considered for incorporation into a vaccine against the SARS-CoV-2. Homology modelling, energy minimization process, and finally Ramachandran model was used for the prediction of a more stable conformation of the spike protein. The most important peptides were screened through the VaxiJen server followed by the IEDB server and CTLPred Score predicted and analysed the desired epitopes. In the final analysis, the peptide EVRQIAPGQTGKIADY (starting from 91) showed the highest antigenicity score (1.3837) as a B-cell epitope although GSTPCNGVEGFNCYFP, starting at 161, showed highest score (0.91) in an initial analysis. On the contrary, as a T-cell epitope, 71 KLNDLCFTNV- 80 was found with the highest antigenicity score (2.6927) which was also found as an epitope in further analysis. A combination of B-cell and T-cell epitopes may evoke a humoral and cell-mediated immune response which will possibly lead to an effective vaccine. Further, the various computational analyses will provide valuable information that will pave the way for modelling a novel vaccine against SARS-CoV-2.
INTRODUCTION
Coronavirus disease 2019 (COVID-19) pandemic is the consequence of the respiratory tract infection caused by novel Coronavirus (2019‐nCoV) which is later known as Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). It has emerged on the eve of 2020 from the Chinese city of Wuhan (Hubei province) [1]. As of June 15, 2020, the pandemic has globally claimed more than 436,900 lives and infected over 8.03 million individuals [2]. Therefore, the development of a vaccine is no longer merely a discussion or part of a debate about whether a vaccine is ultimately needed to suppress the viral spread and to control and prevent SARS-CoV-2. As of June 29, 2020, seventeen candidate vaccines were in clinical evaluation across the globe whereas other 132 candidates in preclinical stages [3].
SARS-CoV-2, which is a positive‐sense single‐stranded RNA virus, has a genome size of approximately 30 kilobases which encode for various structural and non‐structural proteins. The structural proteins of SARS-CoV-2 are the spike (S) protein, the envelope (E) protein, the membrane (M) protein, and the nucleocapsid (N) protein and the non-structural proteins contain open reading frame 1ab (ORF1ab), ORF3a, ORF6, ORF7a, ORF8, and ORF10 [4, 5]. The S protein has two major domains: S1 and S2 [6]. S1 subunit has the receptor-binding domain (RBD) and binds to angiotensin-converting enzyme 2 (ACE2) while S2 mediates the fusion of viral and host cell membranes [7, 8].
The first laboratory-confirmed case in Bangladesh was identified on March 8, 2020 [9]. So far, 90,619 people have been tested positive and 1,209 have died in the country (as of June 15, 2020) [10]. Genomic data of SARS-CoV-2 in Bangladesh is limited, however, efforts from scientists here are underway to make genome sequences available gradually [11]. On May 11, 2020, the first whole-genome sequence (EPI_ISL_437912) from Bangladesh was published. A few days later, the study sequence (EPI_ISL_447590) was made publicly available by the Genome Centre, Jashore University of Science and Technology, Jashore, Bangladesh.
To develop an effective vaccine, the identification of B-cell and T-cell epitopes for SARS-CoV-2 proteins are critical especially for S protein. Both humoral immunity and cellular immunity, provided by B-cell antibodies and T-cell respectively, are essential for effective vaccines [12, 13]. By analyzing an S protein of SARS‐CoV‐2 from Bangladesh, we identified and reported the B-cell and T-cell epitopes. Therefore, the epitopes presented in the study may evoke an effective immune response against SARS‐CoV‐2 for which further experimental analysis will be required. We tend to target the epitopes within the S protein as the protein gave a dominant and long‐lasting immune response against SARS‐CoV that was previously reported [4]. However, our analyses will probably slender down the rummage around for targets for designing a novel vaccine candidate against SARS-CoV-2.
MATERIALS AND METHODS
Nucleotide sequence retrieval and structure prediction
The nucleotide sequence (EPI_ISL_447590) of a Bangladeshi SARS‐CoV‐2 spike (S) glycoprotein was downloaded from the GISAID EpiCoVTM database. The nucleotide sequence was then translated into an amino acid sequence for further analysis. The protein sequence was then applied to form its 3-D model by using SWISS-MODEL [14]. The SMTL ID: 6vyb.1 (https://swissmodel.expasy.org/templates/6vyb.1), chain A, was used as a template that shared 100% sequence identity with the study sequence. The 6vyb.1, chain A, is a SARS-CoV-2 spike ectodomain structure (open state) solved experimentally using Electron Microscopy deposited at early this year in the protein data bank [15]. A PDB file was found after the modeling which was then used for further prediction. Moreover, we minimized energy after homology modeling to find out a stable conformation of proteins using FoldX (http://foldxsuite.crg.eu/). Ramachandran model was also used for finding stable conformation. Ramachandran plot that we used here to determine torsional angles which are permitted, so we could understand the insight into the structure of the peptides, ultimately stable conformation of the peptides. In the Ramachandran plot, the outlier with percentage was measured by SWISS-PDB. From the outliers, the conformations of the phi (Φ) and psi (Ψ) angles were predicted in the protein. Moreover, empirical distribution was observed in this single structure. Ultimately protein stability was confirmed by this value.
Prediction of B-cell epitope
The linear and discontinuous B-cell epitopes based on protein’s 3-D structure were predicted using Ellipro [16]. The prediction parameters were a minimum score and a maximum distance of 0.5 and 6, respectively. The number of residues of each chain was predicted by Ellipro. For identifying B-cell epitopes in an antigen sequence, ABCpred was used to identify the peptide sequence [17]. The protein sequence was submitted where the threshold was 0.51 with a length of 16 to be used for prediction. The antigenicity index of the predicted epitopes was examined and for predicting and designing Interferon-gamma (IFN-γ) inducing peptides, a combination of VaxiJen v2.0 with a threshold of 0.4 and IFN-γ response were used [18, 19].
Prediction of T-cell epitope
The T-cell epitope prediction was performed by using the IEDB tool where all MHC alleles were taken with a threshold of 0.7 for the study [20-22]. The output peptides were again examined by using a combination of Vixen, IFN-γ response, and CTLPred which scores were based on using both support vector machine (SVM) and artificial neural network (ANN) score [23]. In CTLPred, ANN and SVM cut-off scores were used as of 0.51 and 0.36, respectively.
RESULTS
Nucleotide sequence retrieval and structure prediction
The retrieved nucleotide sequence from the GISAID EpiCoVTM database was found to have a length of 694 base pairs. The converted protein sequence was then used for modeling (Figure 1) which showed a 100% sequence identity with template sequence. After conducting the energy minimization process, we got energy minimized state of that protein (Table 1) and a new 3-D figure of the S protein (Figure 2).
From the Ramachandran model (Figure 3), the stable conformation of the PDB file was confirmed with Ramachandran Favoured, and Ramachandran Outliers of 92.58%, and 1.75%, respectively.
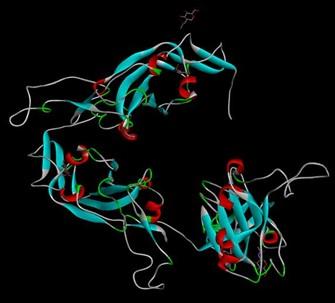
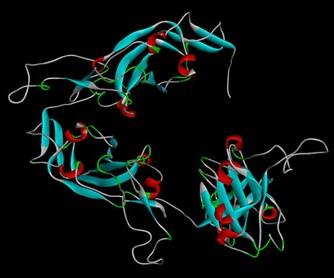
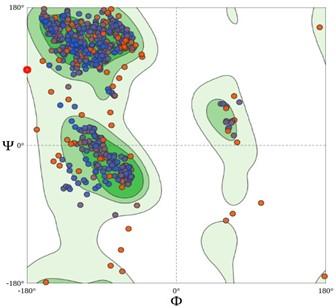
Table 1. Energy minimization of spike protein sequence to find out a stable conformation of protein.
B-cell epitope prediction
The PDB file was used to identify linear and discontinuous epitopes by using Ellipro. This revealed the presence of B-cell epitopes according to their chain ID. It showed 3 chain IDs with their number of residues, 231 equally (Figure 4).
The linear peptides sequence with a threshold of 0.51 provided 23 sequences at a specific length of 16. The antigenicity of peptides was confirmed by the score (Table 2).
For more confirmation, combined processes were done to identify antigen in the spike glycoprotein (Table 3). IFN-γ responses can be either positive or negative depending on the cytokines. The probability of being an antigen of specific peptides was confirmed by VaxiJen with a threshold of 0.4.
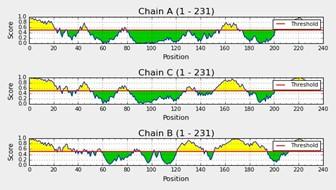
Table 2. Predicted antigenic B-cell linear epitopes found in the spike glycoprotein with their positions and antigenicity scores.
Table 3. Predicted antigenic B-cell linear epitopes found in the spike glycoprotein with their antigenic scores, VaxiJen scores, and IFN-γ responses.
Prediction of T-cell epitope
In spike glycoprotein, a total of 27 MHC-I peptides were predicted at different amino acid positions. However, 12 peptide sequences were found to be antigenic in VaxiJen. IFN-γ responses can be either positive or negative. MHC restriction refers that a particular MHC molecule bounded with the peptides that can be interacted with T cell epitopes. CTLPred score confirms the epitopes probability by combining both support vector machine (SVM) and artificial neural network (ANN) score (Table 4).
Table 4. MHC-I associated antigenic peptide predicted on the spike glycoprotein.
DISCUSSION
To control the current pandemic, the development of new drugs and vaccines is an urgent issue. Especially, effective vaccination or immunotherapy could play a remarkable role in suppressing the viral spread and eventually eliminating it from humans. In the case of vaccine development, a challenge is the absence of sufficient data concerning specific immune responses against SARS‐CoV‐2.
This study has recommended some epitopes that may have the ability to evoke sufficient response in the human body and to be incorporated into a novel vaccine. The 3-D structure of spike (S) protein derived from a Bangladeshi SARS-CoV-2 strain was firstly determined, minimized energy, and confirmed by the Ramachandran model followed by the probable antigenic B-cell, and T-cell epitopes prediction using the amino acid sequence of the same protein.
Recently, a report unveiled that T-cell epitopes are more conducive as a result of more long-lasting immune response mediated by CD8+ T-cell and due to the antigenic drift, by which an antibody is not able to respond against an antibody [24]. In this study, we predicted 12 peptides as T-cell epitopes in a combined strategy (VaxiJen score and IFN-γ responses). All of the peptides were antigenic whereas 71- KLNDLCFTNV- 80 showed the highest antigenicity score (2.6927) though the IFN-γ response was negative which mainly depends on cytokines and can be either positive or negative. In another analysis (CTLPred score), the peptide was also found as an epitope with support vector machine (SVM) and artificial neural network (ANN) score was 0.03 and 0.67438, respectively. Besides these, all peptides were able to interact with the MHC class I alleles.
In the case of SARS‐CoV, antibodies were generated in mice against spike protein that gave protection from the infection [25-27]. Besides, the B-cell epitope provides a strong immune response without causing any adverse effects [28]. Therefore, we also predicted 23 linear B-cell epitopes of 16-mer which could be effective. Among these peptides, the peptide GSTPCNGVEGFNCYFP with start position 161 showed the highest antigenicity score (0.91). But further analysis (VaxiJen score and IFN-γ responses) revealed only 13 peptides and among them, the peptide EVRQIAPGQTGKIADY, started from 91, showed the highest antigenicity score (1.3837) with negative (1) IFN-γ responses.
The potential candidate epitopes, reported in this study, may contribute to the development of a novel vaccine against SARS-CoV-2. A large number of Bangladeshi individuals can be covered by these peptides. The limitation of the study is that all the analyses here were based on a single sequence of SARS‐CoV‐2 derived from Bangladesh. More mutations will be observed as the virus is evolving continuously which may affect our present analysis although most of the mutations are synonymous. However, further experimental analysis will also be required to prove the immunogenicity of the recommended peptides. Overall, the study provides insight that will eventually contribute to the development of a vaccine against SARS‐CoV‐2.
ACKNOWLEDGEMENT
This research received no external funding. We are grateful to the authors and the laboratory from where the study sequence was originated and submitted followed by sharing through the GISAID EpiCoVTM database. Accession ID of the study sequence: EPI_ISL_447590 Originating and submitting laboratory: Genome Centre, Jashore University of Science and Technology, Jashore-7408, Bangladesh. Authors: A. S. M. Rubayet Ul Alam, M. Rafiul Islam, M. Shaminur Rahman, Md. Tanvir Islam, Md. Shazid Hasan, Pravas Chandra Roy, Habiba Ibnat, MD. Ali Ahasan Setu, Tanay Chakrovarty, Sourav Dutta Dip, Ruhul Amin, Md Nur Kabidul Azam, Ovinu Kibria Islam, Hassan M. Al-Emran, Shireen Nigar, Selina Akter, Md. Nazmul Hasan, Iqbal Kabir Jahid, M. Anwar Hossain.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHOR CONTRIBUTIONS
MAI conceived the idea and supervised the project. MH performed the database search. MMRS contributed to perform the experiments. MH analyzed data and wrote the manuscript. MH and MMRS illustrated the figures and prepared the tables. MAI critically revised the manuscript. All the authors proofread and approved the final manuscript.
References
- [1]Wang C, Horby PW, Hayden FG, Gao GF. A novel coronavirus outbreak of global health concern. The Lancet. 2020;395(10223):470-3.
- [2]Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. The Lancet infectious diseases. 2020;20(5):533-4.
- [3]World Health Organization (WHO). DRAFT landscape of COVID-19 candidate vaccines – 29 June 2020. Retrieved June 30, 2020, from https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines.
- [4]Ahmed SF, Quadeer AA, McKay MR. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses. 2020;12(3):254.
- [5]Kiyotani K, Toyoshima Y, Nemoto K, Nakamura Y. Bioinformatic prediction of potential T cell epitopes for SARS-Cov-2. Journal of Human Genetics. 2020;65(7):569-75.
- [6]Ortega JT, Serrano ML, Pujol FH, Rangel HR. Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: An in silico analysis. EXCLI journal. 2020; 19:410.
- [7]Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is 278 Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020. doi: 10.1016/j.cell.2020.02.052.
- [8]Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. European Journal of Internal Medicine. 2020. doi: 10.1016/j.ejim.2020.04.037.
- [9]Hossain I, Khan MH, Rahman MS, Mullick AR, Aktaruzzaman M. The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19) in Bangladesh: A descriptive study. Journal of Medical Science and Clinical Research. 2020;8(04).
- [10]WHO Bangladesh COVID-19 Situation Report #16, 15 June 2020. Retrieved June 18, 2020, from https://www.who.int/bangladesh/emergencies/coronavirus-disease-(covid-19)-update/coronavirus-disease-(covid-2019)-bangladesh-situation-reports.
- [11]Hasan S, Khan S, Ahsan GU, Hossain MM. Genome Analysis of SARS-CoV-2 Isolate from Bangladesh. BioRxiv. 2020. doi:10.1101/2020.05.13.094441.
- [12]Olsson S-E, Villa LL, Costa RL, Petta CA, Andrade RP, Malm C, et al. Induction of immune memory following administration of a prophylactic quadrivalent human papillomavirus (HPV) types 6/11/16/18 L1 virus-like particle (VLP) vaccine. Vaccine. 2007;25(26):4931-9.
- [13]Rappuoli R, Black S, Bloom DE. Vaccines and global health: In search of a sustainable model for vaccine development and delivery. Science Translational Medicine. 2019;11(497): eaaw2888.
- [14]Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic acids research. 2018;46(W1): W296-W303.
- [15]Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020. doi: 10.1016/j.cell.2020.02.058.
- [16]Ponomarenko J, Bui H-H, Li W, Fusseder N, Bourne PE, Sette A, et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC bioinformatics. 2008;9(1):514.
- [17]Saha S, Raghava GPS. Prediction of continuous B‐cell epitopes in an antigen using recurrent neural network. Proteins: Structure, Function, and Bioinformatics. 2006;65(1):40-8.
- [18]Dhanda SK, Vir P, Raghava GP. Designing of interferon-gamma inducing MHC class-II binders. Biology direct. 2013;8(1):30.
- [19]Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647-9.
- [20]Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32(4):511-7.
- [21]Lundegaard C, Lund O, Nielsen M. Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics. 2008;24(11):1397-8.
- [22]Lundegaard C, Nielsen M, Lund O. The validity of predicted T-cell epitopes. Trends in biotechnology. 2006;24(12):537-8.
- [23]Bhasin M, Raghava G. Prediction of CTL epitopes using QM, SVM and ANN techniques. Vaccine. 2004;22(23-24):3195-204.
- [24]Chiou S-S, Fan Y-C, Crill WD, Chang R-Y, Chang G-JJ. Mutation analysis of the cross-reactive epitopes of Japanese encephalitis virus envelope glycoprotein. Journal of general virology. 2012;93(6):1185-92.
- [25]Deming D, Sheahan T, Heise M, Yount B, Davis N, Sims A, et al. Vaccine efficacy in senescent mice challenged with recombinant SARS-CoV bearing epidemic and zoonotic spike variants. PLoS medicine. 2006;3(12).
- [26]Graham RL, Becker MM, Eckerle LD, Bolles M, Denison MR, Baric RS. A live, impaired-fidelity coronavirus vaccine protects in an aged, immunocompromised mouse model of lethal disease. Nature medicine. 2012;18(12):1820.
- [27]Yang Z-y, Kong W-p, Huang Y, Roberts A, Murphy BR, Subbarao K, et al. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature. 2004;428(6982):561-4.
- [28]Rakib A, Sami SA, Islam MA, Ahmed S, Faiz FB, Khanam BH, et al. Epitope-Based Peptide Vaccine Against Severe Acute Respiratory Syndrome-Coronavirus-2 Nucleocapsid Protein: An in silico Approach. BioRxiv. 2020. doi: 10.1101/2020.05.16.100206.