Prevalence and population biology of mastitis-causing Streptococcus uberis using an MLST based approach
Abstract
Streptococcus uberis is a predominant causative agent of both clinical and subclinical varieties of bovine mastitis, an economically significant infection affecting dairy industries around the world. Yet, the genetic and evolutionary relationships among S. uberis strains from different countries are poorly understood. In this study, we used S. uberis’s multilocus sequence typing (MLST) method of genotyping to decipher country-wise prevalence of sequence types (STs) and ST complexes and to delineate genetic relationships among them. Dataset collected from PubMLST database for S. uberis was subjected to clonal cluster and phylogenetic analyses using BURST, globally optimized eBURST (goeBURST) and neighborhood joining algorithm tools, respectively. Whereas certain countries showed clear trends of strain prevalence, others had a more equally weighted, diverse population. Occurrence of different severities of disease varied among countries and displayed no direct correlation with ST. Clonal cluster and phylogenetic analyses predicted the ancestral roles of certain prominent STs and indicated possible strain migration and reticulate and convergent evolution occurring within the S. uberis population at a global scale. Furthermore, genome comparison of selected strains revealed the absence of SUB0822-SUB0826 response regulator proteins from ST-5 and ST-6 indicating their preference for contagious transmission. Information generated from this study would be crucial for monitoring infection outbreaks and directing further genomic investigations.
Keywords
INTRODUCTION
Bovine mastitis is a significant and ubiquitous infectious disease plaguing dairy cattle, thus affecting productivity and profitability of dairy industries worldwide [1–4]. Streptococcus uberis is one of the most commonly identified pathogenic agents responsible for both clinical and subclinical (symptomless) bovine mastitis [3,5,6]. A common constituent of bovine gut flora, S. uberis can become an opportunistic pathogen in hosts with compromised immune systems [7]. Countries, especially those with large scale dairy industries continue to be challenged by mastitis-causing S. uberis, with a recent study estimating up to 6.6% loss in milk yield depending on time and type of infection [2,7–9].
During its first identification in animal beddings in the 1970s, S. uberis was principally classified as an environmental causative agent of bovine mastitis, but since then, several findings have indicated S. uberis to be responsible for both environmental and contagious (cow-to-cow) forms of infection depending on its strain type [7,10,11]. While different risk mitigation strategies such as improved milking practices, post-milking teat disinfection and antimicrobial administration have led to greatly reduced incidences of mastitis derived from contagious pathogens, environmental strains of S. uberis continue to be a major threat owing to their high degree of genetic heterogeneity, diversified pattern of prevalence and pathogenicity [12]. The outcome of genetic heterogeneity within S. uberis populations is that vaccines designed against a particular strain show poor efficacy against others [13,14]. This, coupled with the complex nature of its infection and transmission, makes the development of suitable treatment and control strategies a challenge. Hence, a clear knowledge about country specific strain prevalence is necessary for implementation of effective control measures.
Since mid-2000s, multi locus sequence typing (MLST) has emerged as an efficient method of genotyping pathogens. Over time, different flavors of MLST schemes have evolved, each focusing on different sets of genetic factors such as ribosomal genes (rMLST), core genes (cgMLST) and whole genome (wgMLST) [15–17]. The MLST scheme, originally developed by Coffey et al. in 2006, assigns Sequence Types (STs) to S. uberis isolates based on the allelic profiles of its seven house-keeping genes: carbamate kinase (arcC), D-alanine-D-alanine ligase (ddl), glucose kinase (gki), transketolase (recP), thymidine kinase (tdk), triosephosphate isomerase (tpi) and acetyl-coA acetyltransferase (yqiL), which were selected due to their low propensity towards undergoing mutations [16,18]. Since its development, this MLST scheme has emerged as the gold standard for typing S. uberis isolates, and is widely implemented in research for understanding of S. uberis epidemiology and genetic diversity in local and national outbreaks [8,19–21].
While many studies have focused on S. uberis isolates found in specific countries [7–9], our paper utilizes the MLST scheme to draw inferences on global trends in prevalence patterns and evolutionary relationships among STs and ST complexes, enabling us to understand the population biology of S. uberis-mediated mastitis infections around the globe [16,20]. Information generated from this study will aid in tracking infection outbreak, guiding genomic investigations and thus help in the implementation of country-specific preventive measures.
MATERIALS AND METHODS
Database and prevalence analysis
Publicly available datasets of 1176 S. uberis isolates from 17 countries were collected from University of Oxford’s MLST database, PubMLST (https://pubmlst.org/)[22].The strains were sorted by their source of isolation, infection level in the host (clinical vs subclinical), and country of origin to determine prevalence of STs and ST complexes by country and disease type.
Phylogenetic and clustering analyses
To decipher how STs group into different clonal clusters (ST complexes), the BURST (Based Upon Related Sequence Types) tool of PubMLST was exploited using the default value for group definition whereby STs sharing at least 5 out of 7 housekeeping alleles with one other member of the group were clustered together [23]. To trace the lineage of STs, the clustering software, goeBURST (globally optimized eBURST) was used with a more stringent definition of group whereby strains showing only up to one locus variance i.e. single locus variants (SLV) were clustered together [24]. The in-built neighbor-joining (NJ) algorithm of MLST website’s was used to generate a phylogenetic tree of the 1176 strains, based on the allelic profiles of the seven housekeeping genes used in the MLST scheme, and the resulting tree was visualized using iTOL v3 [25].
In silico analysis of genes and genomes
The number of polymorphic sites and alleles generated from each housekeeping gene was obtained from the polymorphic sites analysis tool of PubMLST where each gene was aligned with a reference sequence from an external database. The genome comparator tool of the server was used to compare strains of different prevalence and infection capacity [18]. The reference strain used was the ST-1 strain, S. uberis 0140J (NCBI Reference Sequence: NC_012004.1) [26]. ST-5 (C6344) and ST-6 (C5072) were chosen to represent highly prevalent strains while the others were less common strains. Some strains collected from subclinical samples of the UK {ST-3 (B190), ST- 25 (C5388) and ST- 11 (AB71)} and Canada {ST- 287 (PE54) and ST- 233 (PE58)} were also compared. All STs selected for comparison belonged to ST complex 5. The SUB0822-SUB0826 gene set was analyzed using STRING v11 (www.string-db.org) to predict protein-protein interactions (PPIs) [27]. For this, the SUB0822-SUB0826 protein sequences derived from NCBI were fed into the server with S. uberis strain 0140J used as the reference organism. The types of interactions were indicated with different colors by the STRING server.
RESULTS
Prevalence pattern of S. uberis STs vary by country and infection level
A total of 1176 S. uberis isolates available on PubMLST were analyzed and sorted by their source, disease state and country of isolation (Table 1). The 1176 isolates were categorized into a total of 587 different STs. The major source of isolates was cattle (n=1016) while the rest were retrieved from different environmental sources such as dirt and animal bedding or from other species such as goat. Inspection of disease background of the isolates revealed that six countries (Canada, Switzerland, Portugal, India, Thailand and China) had only isolates from subclinical mastitis, while the 3 isolates of Denmark were taken from healthy cows. The remaining countries had isolates from both clinical and subclinical varieties of mastitis.
Certain countries showed a visible ST prevalence pattern, with specific STs having higher incidences among that country’s isolates (Figure 1a). For the UK, ST-6, ST-5, ST-20, ST-24 and ST-26 contributed to a significantly large percentage (13%, 10.2%, 9.8%, 4.3% and 3.8%, respectively) of mastitis in the country. Portugal’s three most prevalent STs accounted for ~63% (n = 19) of the country’s disease-causing isolates. On the contrary, some countries such as Sweden and Switzerland harbored a more diverse population of STs. Moreover, no particular strain showed worldwide prevalence. ST-5 and ST-20 for example, although highly prevalent in the UK, were absent in other countries while strains like ST-91 and ST-184 that are commonly found in New Zealand were never isolated in the UK. Figure 1b depicts the frequency of STs by disease background. It can be concluded that no clear correlation appears to exist between a particular strain type and infection level. ST-5 and ST-6 exemplify this, as they have been shown to be prevalent in clinical and subclinical mastitis infections as well as in healthy cattle.
The high number of STs that were generated from 1176 isolates meant that very few strains were shared between two countries. Table 2 is a list of STs that were common between two or more countries. Strains that were shared between countries from two different continents such as ST-233 (shared among UK, Canada and Sweden) give further insights into the evolution and global dissemination of certain S. uberis strains.
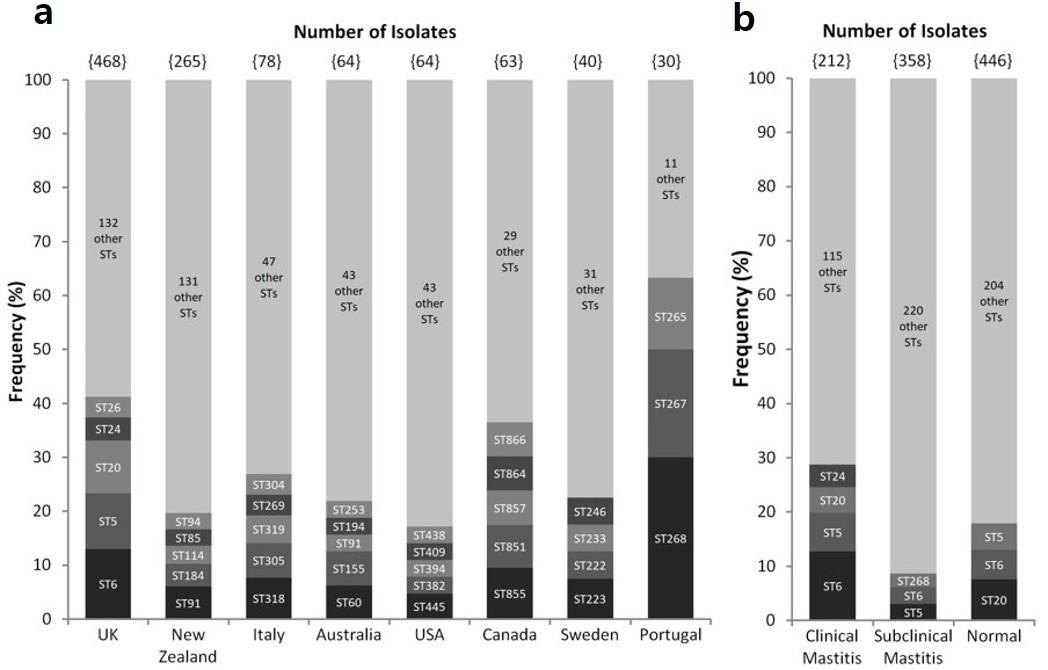
Table 1. Statistical overview of the disease background, source and country of S. uberis isolates collected from PubMLST.
Table 2. List of STs shared between two or more countries.
S. uberis ST-5 and ST-143 complexes are more prevalent in mastitis infections
In the MLST database, strains are either classified into ST-5 complex, ST-86 complex or ST-143 complex. While 35.78% of the STs available in PubMLST were categorized into one of these three groups, a high number of STs were not assigned an ST complex (Table 3). ST-5 complex housed the most prevalent STs in the UK barring ST-20, which probably originated from a different lineage. ST-143 complex on the other hand carried ST-85, ST-91 and ST-184 all of which are prevalent in New Zealand. ST-86 complex accounted for a lesser number of STs and only had one prominent ST (ST-105) which in fact was isolated only from non-mastitis cows. This indicates ST-5 and ST-143 complexes are more dominant producers of mastitis infection in comparison to ST-86 complex.
Table 4 which demonstrates the distribution of bovine isolates from various countries by clinical background and ST complexes further demonstrates that ST complex 86 accounted for a lower number of mastitis cases of both clinical and subclinical varieties.
However, since all three ST complexes had isolates from both clinical and subclinical infections, it can be stated that no direct correlation exists between a type of ST complex and severity of disease. Overall, a large fraction of isolates was not classified into any of the three ST complexes, denoting the existence of other complexes that are yet to be characterized.
Table 3. Number of STs and isolates categorized into ST-5, ST-86 and ST-143 complex.
Table 4. Frequency of strains belonging to three major ST complexes, sorted by disease state and country of isolation.
Clonal clusters originating from different countries share distant variants
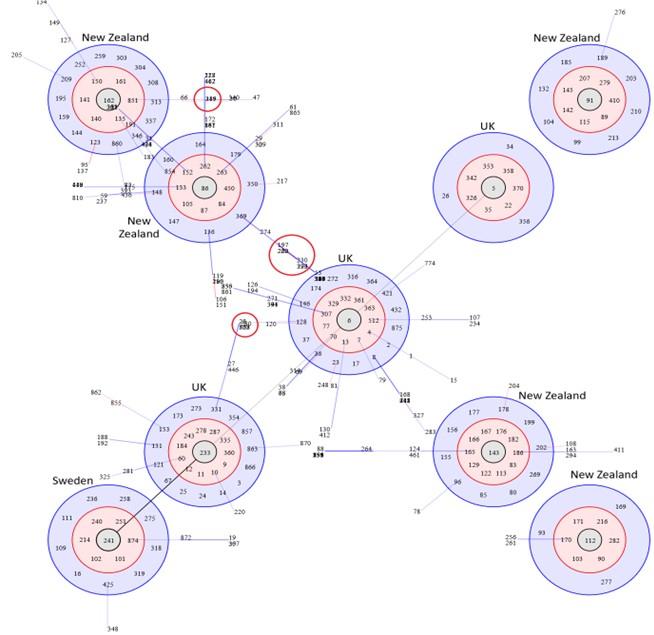
BURST analysis of strains from different countries predicted predominant clones and their ancestral STs. The results revealed isolates from the UK and New Zealand to form multiple clonal complexes with a central or primitive ST: the UK isolates formed three clonal complexes with ST-5, ST-6 and ST-233 at the center, while New Zealand formed five clonal complexes (the highest for any country) with ST-86, ST-91, ST-112 and ST-143 at the center (Figure 2). The ST-241 complex belonged to Sweden. Other countries did not form any clonal complex of their own and therefore were not depicted in Figure 2. Lines joining ST-5, ST-6 and ST-233 complexes of the UK support the common lineage shared by strains under these three clonal complexes, while ST-233 complex connecting with ST-241 from Sweden demonstrates cross-country dissemination of strains. In contrast, only two clonal complexes from New Zealand (ST-86 and ST-162 clusters) formed a linkage. BURST output also revealed that triple locus variants (TLVs) are sometimes shared between clonal clusters originating from two different geographical regions (Figure 4); ST-6 complex and ST-86 complex, predominant in the UK and New Zealand, respectively, share common distant variants. This observation combined with possible strain migration over history, underpins the likelihood of reticulate as well as convergent evolution occurring within S. uberis population worldwide.
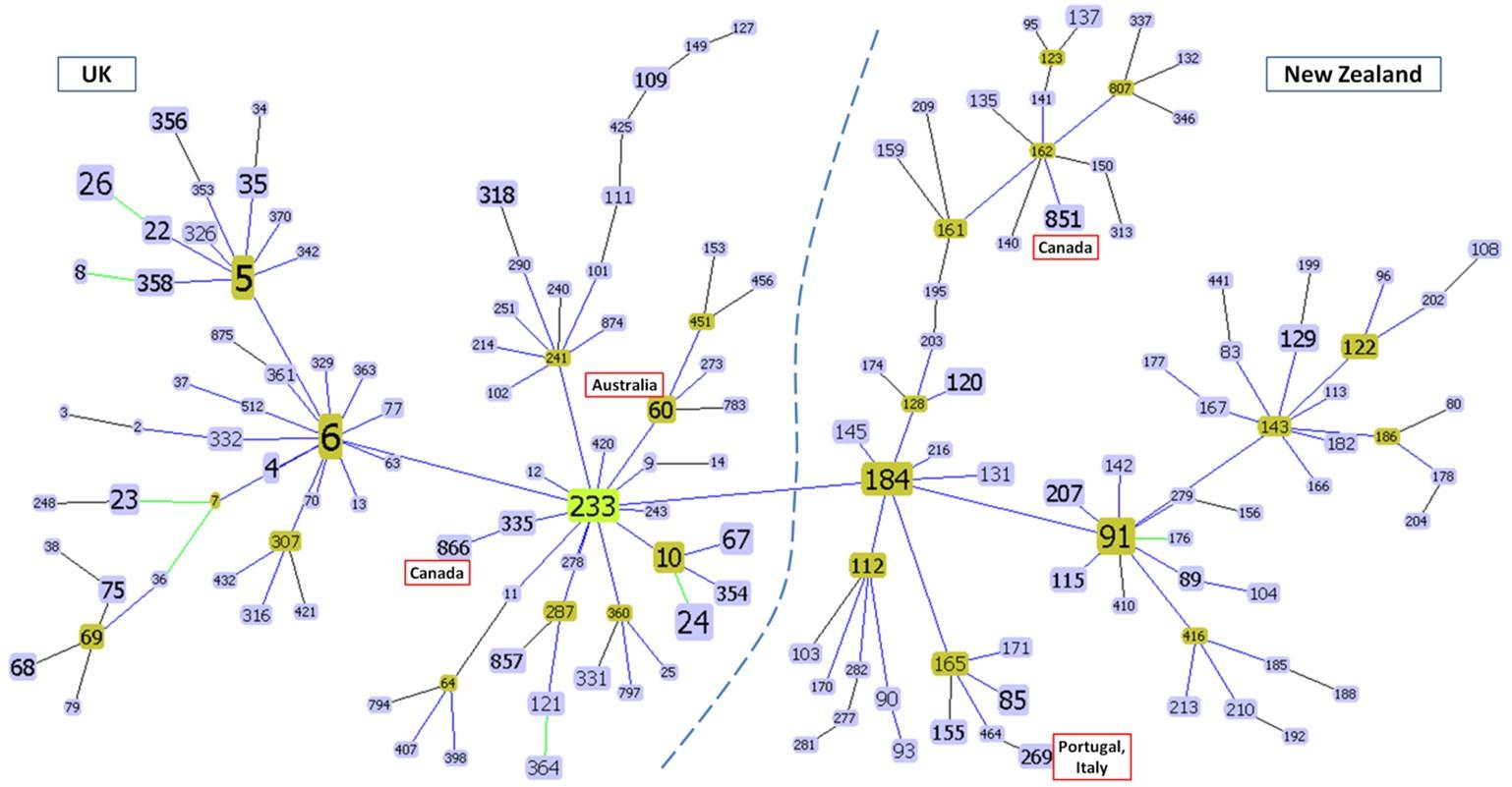
The goeBURST results predicted evolutionary linkage between different ST complexes (Figure 3). The primary chain of clusters consisted of ST-6, ST-5 and ST-233 complexes on one side and ST-184, ST-91 and ST-143 complexes on the other. This created two topologically distinct zones of clusters, one containing STs commonly found in the UK and other European countries and another containing STs predominant in the Oceanic Countries- New Zealand and Australia (Figure 3). Some anomalous clustering trends such as ST-60 of Australia branching from the European ST-233 complex and ST-851 of Canada clustering with Oceanic STs were also observed in the tree.
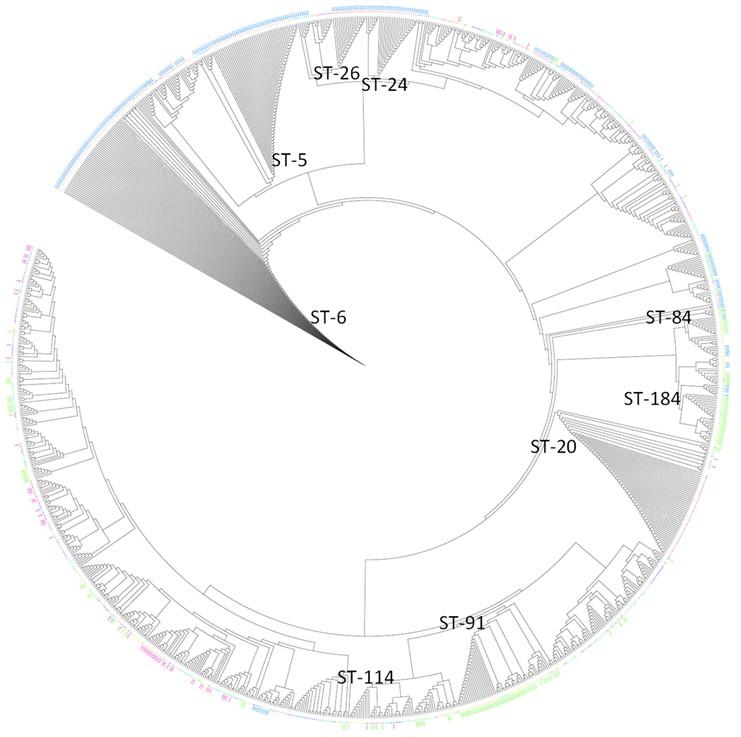
Certain prevalent S. uberis STs are ancestral to other STs
The NJ tree (Figure 4) exhibited interspersing of strains from different regions and thus no clear divide could be visualized between ST groups of different countries. The positions of highly prevalent STs from the UK (ST-5, ST-6, ST-20, ST-24 and ST-26) and New Zealand (ST-84, ST-91, ST-114 and ST-184) exhibit this. ST-20 for example is situated farther from the UK’s other prevalent STs, thus again denoting its different origin from other ST-5 complex strains. ST-6, ST-5 and ST-20 are positioned closer to the root in comparison to other strains indicating their primitive nature (Figure 4).
Genome comparison showed prevalent strains to be devoid of regulatory genes
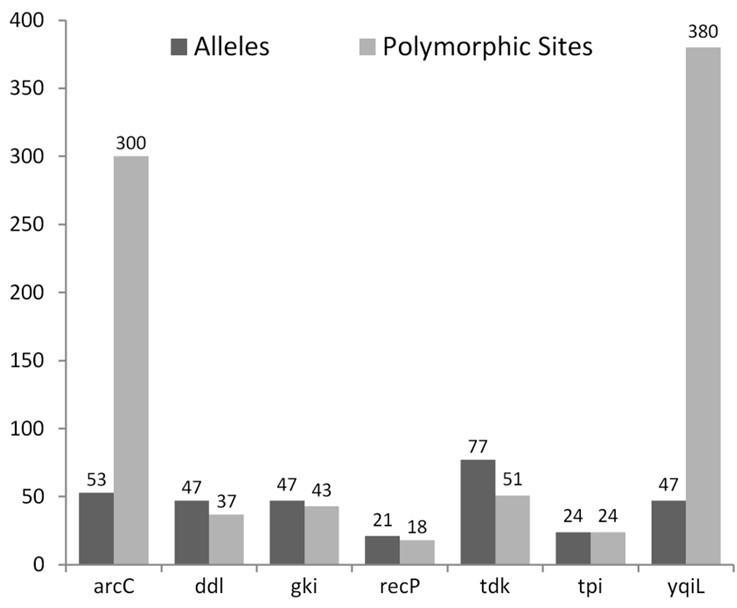
Analysis of polymorphic sites within each of the seven housekeeping genes showed arcC and yqiL carrying disproportionately high numbers of polymorphic sites with far fewer numbers of alleles, thus indicating simultaneous occurrence of multiple mutations in any given allele (Figure 5). The other five genes, in comparison, had allele numbers greater than or equal to the number of polymorphic sites present in them.
Genomic comparison of the eight aforementioned strains revealed that ST-5 and ST-6 did not carry a set of genes (SUB0822, SUB0823, SUB0824/malP, SUB0825 and SUB0826) which is involved in regulating a cell’s response to environmental changes. Genome comparison amongst clinical STs and subclinical STs of UK and Canada did not point towards any genetic markers that might be associated with disease states. However, high variability (i.e., loci showing different allele types for each strain) was observed for many proteins related to replication (e.g. DNA gyrase and topoisomerases) as well as for those associated with a wide range of metabolic processes.
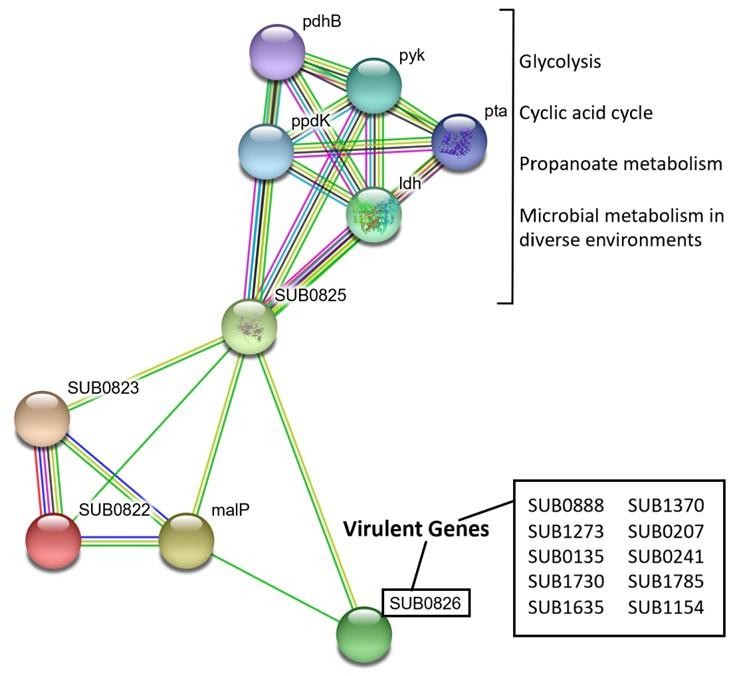
SUB0822-SUB0826 form a valid PPI network
The string server showed a network of protein interactions with their function (Figure 6). The green and yellow lines between SUB0826 and SUB0825 and SUB0823 represent interactions based on gene neighborhood and text mining. The PPI network thus clearly showed that SUB0822-SUB0826 generate proper signals via response regulator (SUB0822) and sensor kinase (SUB0823) which are necessary to function sodium-dependent malate transporter (SUB0824/malP) and NAD-dependent malic enzyme (SUB0825) which needed to produce NADH. SUB0825 interacts with another set of proteins: Idh, ppdK, pdhB, pyk and pta. Combinations of these proteins are involved in glycolysis, citric acid cycle and propanoate metabolism and a subset of them (ldh, ppdK, pyk and pta) have been confirmed to control microbial metabolism in diverse environments. Several virulence proteins of S. uberis have been identified in recent studies [24]. Significant interactions between SUB0822-SUB0826 proteins and virulent proteins were not observed except SUB0826 (putative surface-anchored subtilase family protein) (Figure 6).
DISCUSSION
Bovine pathogens like S uberis pose a great threat to the economy of dairy industries worldwide and outbreak of this detrimental organism could cause thousands of dairy farmers to incur devastating losses. Therefore, it is essential to study the origins of S. uberis strains and to monitor their clonal propagations periodically. As shown in this study, MLST can be a useful tool for dissecting prevalence patterns, and evolutionary history of S. uberis strains. Such studies could provide crucial information for monitoring disease outbreaks and would aid in driving large-scale comparative genomic studies, the results of which could be utilized to design more discriminative and effective treatments.
Our finding of highly prevalent STs within mastitis infections in the UK and Canada is consistent with recent findings showing the same STs to be more frequent in infected cattle of the respective countries [7,28].. As most other countries showed a far more diverse population of STs, this geographical difference in prevalence patterns is indicative of different mechanisms of infection transmission. Studies in the UK have previously revealed that cattle within the same herd tend to carry one particular ST [7,10] indicating contagious transmission and explaining high incidences of certain strains over others in cows. On the contrary, strains from countries that have a more genetically diverse population of S. uberis likely infect cattle from environmental reservoirs such as pastures and animal beddings [29].
Since no apparent correlation could be established between disease severity (clinical vs subclinical) and ST or ST complex, this study further confirms the combinatorial effect of strain genetics and confounding factors (such as environmental and host factors), on disease outcome. A recent finding demonstrated that Indian strains ST-439 and ST-475 derived from subclinical infections elicit different inflammatory responses in bovine hosts, demonstrating that immune responses can largely vary from one strain to another at any given infection level [30].
BURST and goeBURST analyses emphasized the likelihood of the UK’s most dominant strains sharing a common lineage whereas some of New Zealand’s clonal clusters could have emerged from several distinct ancestors. In addition, the presence of common TLVs between ST complexes originating from two different continents underpins the possibility of the occurrence of convergent evolution within S. uberis population across the world, whereby selection of beneficial genes/alleles as a result of environmental pressures has led to the emergence of strains that can be evolutionarily linked to ancestors from different geographical regions. Despite major ST complexes of two different continents exhibiting spatial separation, the clustering of foreign isolates such as those from Canada with both European and Oceanic clonal clusters, as observed in goeBURST analysis, underpin the complex evolutionary forces as mentioned above. Strain migration and subsequent gathering of mutations could also explain this genetic relatedness to geographically separated groups of strains.
Comparative genomics approaches have revealed diverse virulence factors in S. uberis [31]. The SUB0822-SUB0826 family of genes that were absent from the UK’s prevalent STs formed a valid PPI network, indicating that they are both genetically and functionally related. Interactions were predicted by gene neighborhood, and thus it can be concluded that all the genes in the network are topologically proximate, while the interactions predicted by text mining depict potential protein-protein binding sites. As these proteins have been characterized to be essential for adapting to environmental changes, it is possible that their absence renders ST-5 and ST-6 incapable of surviving outside the host system, making them reliant on cow-to-cow transmission. On the contrary, STs possessing this machinery can adapt to changing environments outside the host and can therefore be held in environmental reservoirs.
The lack of any stark difference in genetic content between the subclinical and clinical strains, however, further reinforces the complex nature of S. uberis pathogenesis and the involvement of other confounding factors in determining disease severity. Nevertheless, because of having wgMLST information of only a few isolates available, it is difficult to draw a definite conclusion regarding the significance of this set of proteins in dictating transmission route. In future, large scale comparative genomics studies are needed to excavate insights regarding the importance of gene sets that are vital for the virulence, fitness and transmission of S. uberis from both environmental and bovine reservoirs.
CONCLUSION
The results of this study demonstrate that the MLST method of genotyping and clonal cluster analyses of STs can effectively predict prevalence, evolution and migration activity of S. uberis isolates. Information from these results can produce inferences regarding transmission route of strains, altogether resulting in the implementation of more discriminative control strategies to prevent future outbreaks. In conclusion, it is pertinent to pave way for large scale genomic studies to draw any statistically significant results regarding the association of a gene with a particular mode of infection (environmental or contagious) and disease severity (clinical or subclinical). In addition to that, structural analyses of proteins encoded by these relevant genes may aid in the design and development of treatments to combat clinical and subclinical mastitis in different geographical regions.
ACKNOWLEDGEMENT
No specific funds were received for this study. The manuscript does not contain clinical studies or patient data.
AUTHOR CONTRIBUTIONS
AR and AB were involved in conceptualization and design of the study and original draft preparation. AR, AB and TT did the data analysis. AR and MAI edited the draft. MH supervised the study and reviewed the paper. All authors have read and approved the final version of the manuscript.
CONFLICTS OF INTEREST
There is no conflict of interest among the authors.
References
- [1]Halasa T, Huijps K, Østerås O, Hogeveen H. Economic effects of bovine mastitis and mastitis management: A review. Vet Q 2007;29:18–31.
- [2]Heikkilä AM, Liski E, Pyörälä S, Taponen S. Pathogen-specific production losses in bovine mastitis. J Dairy Sci 2018;101:9493–504.
- [3]Keane OM. Symposium review: Intramammary infections—Major pathogens and strain-associated complexity. J Dairy Sci 2019;102:4713–4726.
- [4]Aghamohammadi M, Haine D, Kelton DF, Barkema HW, Hogeveen H, Keefe GP, et al. Herd-Level Mastitis-Associated Costs on Canadian Dairy Farms. Front Vet Sci 2018;5:100.
- [5]Zadoks RN, Middleton JR, McDougall S, Katholm J, Schukken YH. Molecular Epidemiology of Mastitis Pathogens of Dairy Cattle and Comparative Relevance to Humans. J Mammary Gland Biol Neoplasia 2011;16:357–372.
- [6]Jayarao BM, Gillespie BE, Lewis MJ, Dowlen HH, Oliver SP. Epidemiology of Streptococcus uberis intramammary infections in a dairy herd. J Vet Med Ser B 1999;46:433–442.
- [7]Davies PL, Leigh JA, Bradley AJ, Archer SC, Emes RD, Green MJ. Molecular Epidemiology of Streptococcus uberis Clinical Mastitis in Dairy Herds: Strain Heterogeneity and Transmission. J Clin Microbiol 2016;54:68–74.
- [8]Reyes Vélez J, Cameron M, Rodríguez-Lecompte JC, Xia F, Heider LC, Saab M, et al. Whole-Genome Sequence Analysis of Antimicrobial Resistance Genes in Streptococcus uberis and Streptococcus dysgalactiae Isolates from Canadian Dairy Herds. Front Vet Sci 2017;22:63.
- [9]Vezina B, Al-harbi H, Ramay HR, Soust M, Moore RJ, Olchowy TWJ, et al. Sequence characterisation and novel insights into bovine mastitis-associated Streptococcus uberis in dairy herds. Sci Rep 2021;11:1–16.
- [10]Zadoks RN, Gillespie BE, Barkema HW, Sampimon OC, Oliver SP, Schukken YH. Clinical, epidemiological and molecular characteristics of Streptococcus uberis infections in dairy herds. Epidemiol Infect 2003;130:335–349.
- [11]Wente N, Klocke D, Paduch JH, Zhang Y, Seeth M tho, Zoche-Golob V, et al. Associations between Streptococcus uberis strains from the animal environment and clinical bovine mastitis cases. J Dairy Sci 2019;102:9360–9369.
- [12]Klaas IC, Zadoks RN. An update on environmental mastitis: Challenging perceptions. Transbound Emerg Dis 2018;65:165–188.
- [13]Finch JM, Winter A, Walton AW, Leigh JA. Further studies on the efficacy of a live vaccine against mastitis caused by Streptococcus uberis. Vaccine 1997;15:1138–1143.
- [14]Collado R, Montbrau C, Sitjà M, Prenafeta A. Study of the efficacy of a Streptococcus uberis mastitis vaccine against an experimental intramammary infection with a heterologous strain in dairy cows. J Dairy Sci 2018;101:P10290-10302..
- [15]Perez-Losada M, Porter ML, Viscidi RP, Crandall KA. Multilocus Sequence Typing of Pathogens. Genet Evol Infect Dis 2011:503–521.
- [16]Coffey TJ, Pullinger GD, Urwin R, Jolley KA, Wilson SM, Maiden MC, et al. First Insights into the Evolution of Streptococcus uberis: a Multilocus Sequence Typing Scheme That Enables Investigation of Its Population Biology. Appl Environ Microbiol 2006;72:1420–1428.
- [17]Pullinger GD, López-Benavides M, Coffey TJ, Williamson JH, Cursons RT, Summers E, et al. Application of Streptococcus uberis multilocus sequence typing: Analysis of the population structure detected among environmental and bovine isolates from New Zealand and the United Kingdom. Appl Environ Microbiol 2006;72:1429–1436.
- [18]Jolley KA, Maiden MCJ. BIGSdb : Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 2010;11:595.
- [19]Käppeli N, Morach M, Zurfluh K, Corti S, Nüesch-Inderbinen M, Stephan R. Sequence types and antimicrobial resistance profiles of streptococcus uberis isolated from bovine mastitis. Front Vet Sci 2019;6:234.
- [20]Phuektes P, Mansell PD, Dyson RS, Hooper ND, Dick JS, Browning GF. Molecular Epidemiology of Streptococcus uberis Isolates from Dairy Cows with Mastitis. J Clin Microbiol 2001;39:1460–1466.
- [21]Shome BR, Bhuvana M, Mitra S Das, Krithiga N, Shome R, Velu D, et al. Molecular characterization of Streptococcus agalactiae and Streptococcus uberis isolates from bovine milk. Trop Anim Health Prod 2012;44:1981–1992.
- [22]Jolley KA, Bray JE, Maiden MCJ. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res 2018;3.
- [23]Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol 2004;186:1518–30.
- [24]Francisco AP, Bugalho M, Ramirez M, Carriço JA. Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach 2009;10:152.
- [25]Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 2016;44:W242–W245.
- [26]Ward PN, Holden MTG, Leigh JA, Lennard N, Bignell A, Barron A, et al. Evidence for niche adaptation in the genome of the bovine pathogen Streptococcus uberis. BMC Genomics 2009;10:54.
- [27]Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47:607–613.
- [28]Reyes Velez J, Rodriguez-Lecompte JC, Sanchez J, Zadoks R. Use of whole genome sequence to identify new sequence types of Streptococcus uberis from dairy cattle, 2017. 4: 63
- [29]Wald R, Baumgartner M, Gutschireiter J, Bazzanella B, Lichtmannsperger K, Wagner M, et al. Comparison of the population structure of Streptococcus uberis mastitis isolates from Austrian small-scale dairy farms and a Slovakian large-scale farm. J Dairy Sci 2020;103:1820–1830.
- [30]Mitra S Das, Shome BR, Bhuvana M, Velu D, Banerjee A, Bankar K, et al. Streptococcus uberis ST439 and ST475 induce differential inflammatory responses in a mouse intramammary infection model. Gene 2016;585:247–255.
- [31]Hossain M, Egan SA, Coffey T, Ward PN, Wilson R, Leigh JA, et al. Virulence related sequences; insights provided by comparative genomics of Streptococcus uberis of differing virulence. BMC Genomics 2015;16:334.